electroencephalography detailed
Reading File
Finding Sources
Searching the Web
electroencephalography EEG principles clinical applications 2024
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Finding Sources
Reading File
Searching PubMed
electroencephalography clinical applications review
I now have comprehensive, rich content from multiple authoritative textbooks. Let me compile the full detailed answer.
Electroencephalography (EEG) - A Detailed Overview
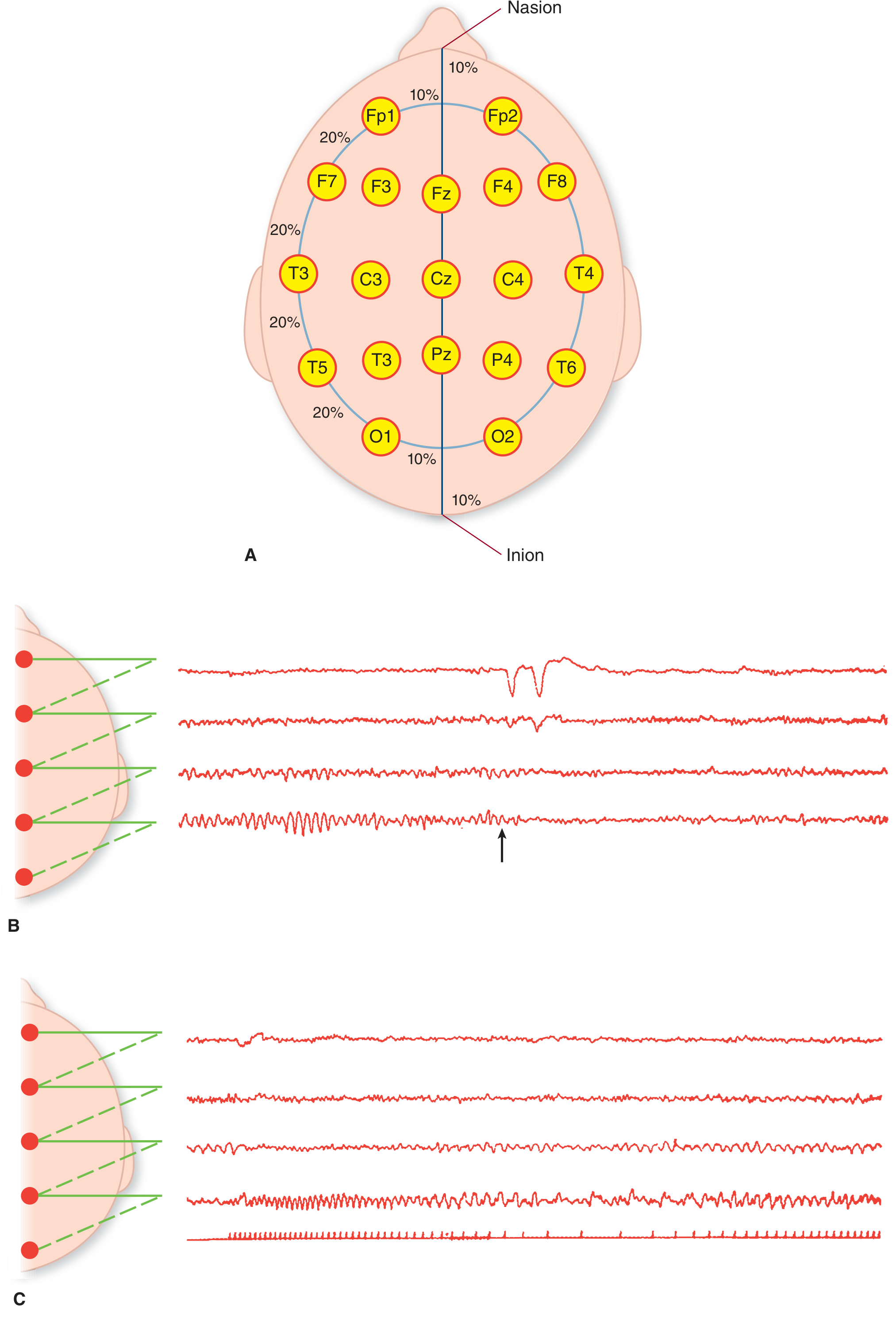
Figure: The International 10-20 electrode placement system (top) and representative EEG channel tracings - Adams and Victor's Principles of Neurology, 12th Ed.
1. History and Basic Concept
The electroencephalograph was developed by Hans Berger in 1929. It records the spontaneous electrical activity generated in the cerebral cortex, which reflects the summated effects of innumerable excitatory and inhibitory synaptic potentials upon cortical neurons - not action potentials themselves, but postsynaptic currents flowing in the extracellular space.
This cortical activity is strongly influenced and synchronized by subcortical structures, particularly the thalamus and the high brainstem reticular formation. Efferent impulses from these deep structures entrain cortical neurons to produce characteristic rhythmic brain-wave patterns (alpha rhythm, sleep spindles). Alpha waves will not occur in the cerebral cortex without cortical connections with the thalamus.
Because surface EEG sums only synchronous activity, strong nonsynchronous signals cancel each other out - this is why opening the eyes (which activates millions of asynchronous neurons) paradoxically produces lower-voltage, irregular beta waves rather than higher voltage.
- Adams and Victor's Principles of Neurology, 12th Ed.
- Eric Kandel - Principles of Neural Science, 6th Ed.
2. Technical Setup
Electrodes
- Silver or silver-silver chloride discs, ~0.5 cm in diameter, placed on the scalp with a conductive medium
- Modern EEG machines: 8 to 32+ amplifying channels recording simultaneously
- Frequency range displayed: 0.5 to 30 Hz at standard paper speed of 3 cm/s
- Amplitude range: 20 to 100 µV typically
- Signal displayed as voltage-versus-time; negative voltage deflects upward, positive downward by convention
The International 10-20 System
The dominant electrode placement scheme uses 19 active electrodes + reference/ground placed at intervals of 10% or 20% of the hemi-circumference of the head. Electrode names correspond to underlying brain regions:
- Fp - frontopolar
- F - frontal
- C - central
- P - parietal
- O - occipital
- T - temporal
- z suffix - midline electrodes (Fz, Cz, Pz)
- Odd numbers = left hemisphere; even numbers = right hemisphere
Montages
A montage is a specific configuration of electrode pairs recorded together. Two main types:
- Bipolar montage: each channel records the voltage difference between two adjacent electrodes - helps localize focal abnormalities
- Referential (common reference) montage: each electrode is compared to a common reference point - better for amplitude comparison across the scalp
3. Normal EEG Patterns
The Four Core Rhythms
| Rhythm | Frequency | Amplitude | Location | State |
|---|---|---|---|---|
| Alpha (α) | 8-13 Hz | ~50 µV | Occipital, parietal | Eyes closed, relaxed wakefulness |
| Beta (β) | >13 Hz (up to 80 Hz) | 10-20 µV | Frontal, parietal | Alert, eyes open, active mental work |
| Theta (θ) | 4-7 Hz | Variable | Parietal, temporal | Drowsiness, early sleep; abnormal if prominent in awake adult |
| Delta (δ) | <3.5 Hz | 50-350 µV | Diffuse | Deep sleep, infancy; always abnormal in awake adult |
Gamma (γ) waves (>30 Hz) are also recognized and associated with high-level cognitive processing, though less routinely reported in clinical EEGs.
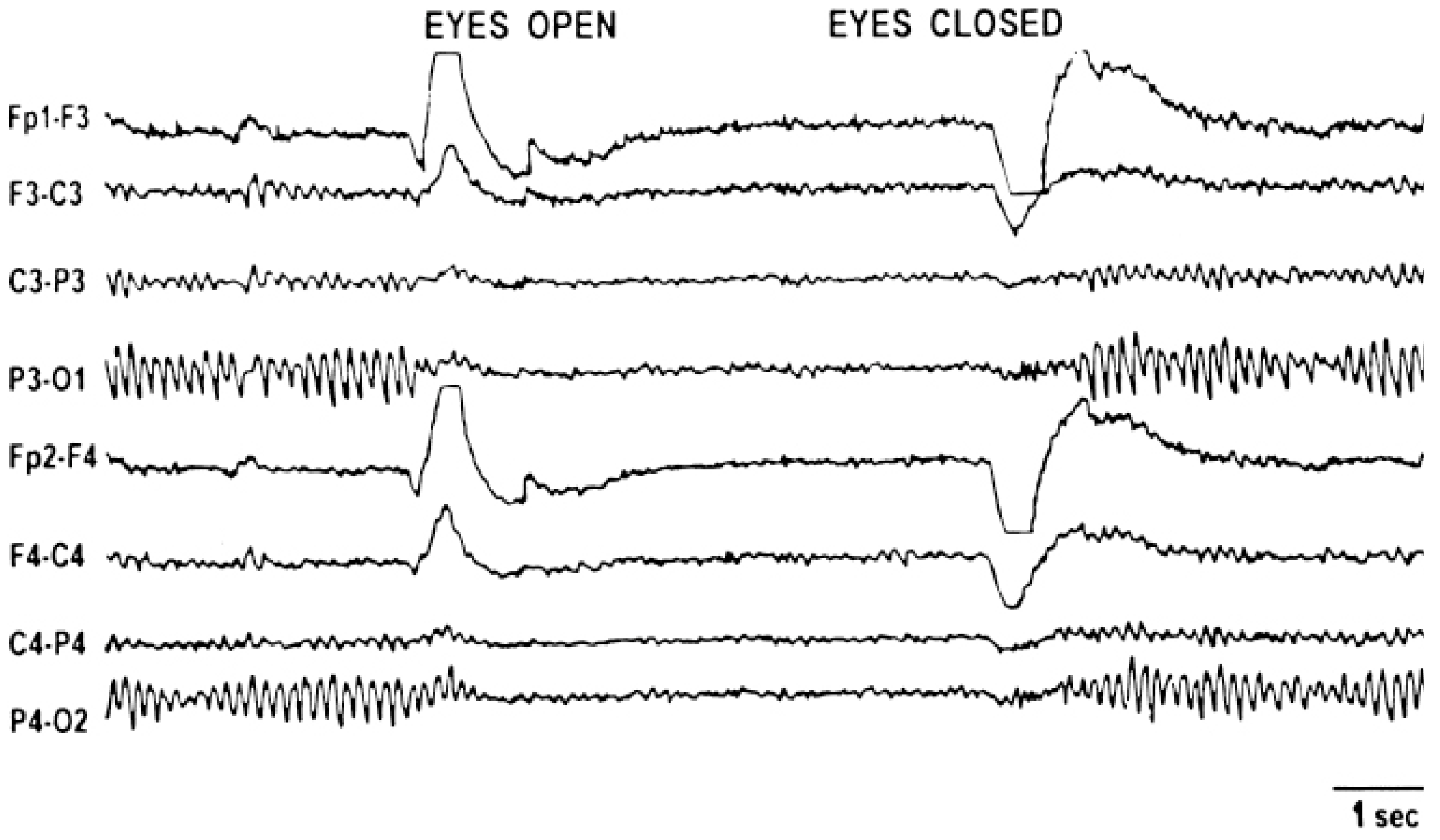
Figure: Normal EEG demonstrating alpha rhythm (P3-O1, P4-O2 leads) abolished on eye opening, replaced by low-voltage beta activity - Kaplan & Sadock's Synopsis of Psychiatry
Key properties of normal alpha:
-
Most intense in the occipital region
-
Waxes and wanes spontaneously
-
Attenuated or suppressed by eye opening or mental effort ("alpha blocking")
-
Frequency is almost invariant for a given individual, though it slows with aging
-
Benzodiazepines and sedating drugs increase beta-frequency activity prominently
-
Guyton and Hall Textbook of Medical Physiology
-
Adams and Victor's Principles of Neurology, 12th Ed.
4. Sleep EEG
EEG patterns change dramatically across sleep stages:
| Stage | EEG Features |
|---|---|
| Relaxed wakefulness | Posterior alpha rhythm |
| Drowsiness (NREM Stage 1) | Alpha drops out, replaced by irregular low-voltage theta |
| Light sleep (NREM Stage 2) | Sleep spindles (14 Hz, 1-2 second bursts = sigma waves) + K-complexes (large biphasic complexes at vertex) |
| Deep sleep (NREM Stage 3/4) | High-voltage delta waves predominate (slow-wave sleep) |
| REM sleep | Low-voltage, mixed-frequency, similar to wakefulness; sawtooth waves may appear |
Sleep spindles are generated by thalamocortical circuits. Vertex sharp waves are seen at central electrode sites (Cz), particularly in younger persons, as drowsiness deepens.
EEG during sleep deprivation or during natural/sedated sleep can activate paroxysmal EEG discharges that are not apparent in the routine awake tracing - making sleep EEG an important part of epilepsy workup.
- Kaplan & Sadock's Synopsis of Psychiatry
- Costanzo Physiology, 7th Ed.
5. Activating Procedures
During routine EEG recording, several provocative maneuvers are used to bring out abnormalities:
-
Hyperventilation: 20 breaths/min for 3 minutes - lowers pCO₂, causes cerebral vasoconstriction, enhances absence seizure patterns (3 Hz spike-and-wave). Normal children may show "buildup" (delta) activity during HV, which stops after.
-
Photic stimulation (intermittent phased light): flashing strobe at various frequencies (1-30 Hz). A photic driving response is normal. Abnormal responses include:
- Photomyoclonic response: myoclonic jerks during stimulation
- Photoparoxysmal response: epileptiform discharges that outlast the stimulus
- Photoconvulsive response: full seizure triggered; seen in alcohol/sedative withdrawal
-
Sleep deprivation: 24 hours of deprivation can activate paroxysmal discharges in susceptible patients
-
Sleep (natural or sedated): widens the recording window for epileptiform activity
6. Abnormal EEG Patterns
Focal Slowing
- Delta waves (1-3 Hz, focal): indicate a destructive cortical lesion - tumor, infarct, abscess, subdural hematoma, encephalitis
- Theta waves (4-7 Hz, focal or diffuse): less severe but still pathological when prominent during wakefulness
- Large acute middle cerebral artery infarctions produce large areas of focal slowing; lacunar or brainstem infarcts often leave the surface EEG normal despite clinical deficits
Generalized Slowing
Indicates diffuse cerebral dysfunction:
- Metabolic encephalopathies: uremia, hepatic coma, hypoglycemia, anoxia, hypercapnia
- Severity of slowing generally correlates with depth of impaired consciousness
- Triphasic waves (bilaterally synchronous high-amplitude sharp triphasic waves, frontally predominant): characteristic of hepatic encephalopathy (also seen in renal/pulmonary failure, acute hydrocephalus)
Epileptiform Patterns
- Spike: transient high-voltage waveform, pointed peak, duration 20-70 ms
- Sharp wave: similar morphology, duration 70-200 ms
- Spike-and-wave complex: 3 Hz = typical absence epilepsy (petit mal)
- Polyspike-and-wave: myoclonic epilepsy syndromes
- Focal spikes/sharp waves: focal (partial) epilepsy
Importantly:
- In 30% of patients with absence epilepsy and 50% with generalized tonic-clonic epilepsy, a single interictal EEG is normal
- 30-40% of epilepsy patients have nonspecifically abnormal records between seizures
- Antiepileptic drugs can mask interictal abnormalities
- A completely normal EEG during a clinical convulsion strongly suggests psychogenic nonepileptic seizure (PNES)
Burst-Suppression
- Alternating periods of EEG activity (bursts of sharp/irregular delta) and flat isoelectric periods
- Seen with severe anoxic brain injury, deep anesthesia, severe metabolic derangement
Electrocerebral Silence (Isoelectric EEG)
- Complete absence of EEG activity
- Component of the diagnosis of brain death, but must be interpreted carefully:
- Can be mimicked by deep sedation with barbiturates, profound hypothermia, or hypothyroid coma
- Not sufficient alone for brain death determination - clinical criteria must also be met
Periodic Discharges
- Periodic sharp wave complexes at 1-3/second in temporal regions: strongly suggest herpes simplex encephalitis
- Periodic bisynchronous sharp wave bursts: almost pathognomonic of Creutzfeldt-Jakob (prion) disease
- SSPE (subacute sclerosing panencephalitis): also shows periodic high-amplitude sharp wave bursts
Alpha Coma
- EEG shows apparent 8-12 Hz alpha-like activity distributed broadly (not just posteriorly)
- Different from normal alpha - slightly variable in frequency
- Usually a transitional pattern after global anoxia, or with large acute pontine lesions; carries poor prognosis
Breach Rhythm
-
Focal fast (beta) activity over a skull defect (bone acts as a high-frequency filter; its absence allows abundant cortical fast activity to pass through)
-
Adams and Victor's Principles of Neurology, 12th Ed.
-
Kaplan & Sadock's Comprehensive Textbook of Psychiatry
7. Clinical Applications
Epilepsy
The primary and most important use of EEG. It classifies seizure type, guides antiepileptic drug selection, evaluates surgical candidacy, and monitors treatment response.
| Seizure Type | EEG Pattern |
|---|---|
| Absence (petit mal) | 3 Hz generalized spike-and-wave, abrupt onset/offset |
| Juvenile myoclonic epilepsy | Polyspike-and-wave, faster |
| Generalized tonic-clonic | Generalized spike-and-wave, evolving polyspike |
| Infantile spasms (West syndrome) | Hypsarrhythmia (chaotic high-amplitude activity) |
| Lennox-Gastaut syndrome | Slow (2-2.5 Hz) spike-and-wave, fast bursts during sleep |
| Focal seizures | Rhythmic focal discharges at ictal onset zone |
| Benign childhood epilepsy with centrotemporal spikes (BECTS) | Focal centrotemporal/rolandic spikes, sleep-activated |
Encephalopathy Assessment
- Quantifies severity and tracks progression of metabolic, toxic, and inflammatory brain diseases
- Useful in distinguishing delirium from psychiatric disease (normal EEG in functional/psychiatric states)
Coma Evaluation
- Differentiates nonconvulsive status epilepticus (NCSE) from other causes of impaired consciousness
- Grading of coma severity
- Prognostication after cardiac arrest (burst suppression or flat EEG = poor prognosis)
Sleep Medicine
- Polysomnography uses EEG together with EOG, EMG, and respiratory sensors to characterize sleep architecture
- Diagnoses REM sleep behavior disorder, narcolepsy, nocturnal epilepsy
Intraoperative Monitoring
- Monitors cerebral perfusion during carotid endarterectomy and cardiac surgery
- Processed EEG (bispectral index [BIS], others) used to assess depth of anesthesia
Specific Diseases
- Herpes simplex encephalitis: periodic temporal sharp waves at 1-3/second - supports early treatment decision
- Creutzfeldt-Jakob disease: periodic bisynchronous sharp wave complexes - important adjunctive test
- Hepatic encephalopathy: triphasic waves
- Brain death determination: isoelectric EEG as ancillary test
EEG-Based Brain-Computer Interfaces (BCI)
A rapidly growing field where EEG signals are decoded in real-time to allow communication or device control in paralyzed patients. Recent 2025 review in Military Medical Research summarizes applications in motor rehabilitation, communication, and neuromodulation (PMID: 40128831).
8. Limitations
- Spatial resolution is poor: scalp EEG averages activity from millions of neurons and cannot record from deep structures (hippocampus, amygdala, thalamus, brainstem) directly
- Deep foci (medial temporal, orbital frontal) may produce seizures without visible scalp EEG changes
- Temporal resolution is excellent (millisecond scale)
- A normal interictal EEG does not exclude epilepsy - up to 50% of grand mal epilepsy patients have a normal single EEG
- Interpretation requires significant expertise; artifacts (movement, ECG, eye blink) can mimic pathology
- EEG has largely been replaced by MRI/CT for structural lesion localization
9. Variants and Extended Techniques
| Technique | Description |
|---|---|
| Ambulatory EEG | Portable recording over 24-72 hours; increases diagnostic yield |
| Video-EEG | Simultaneous video and EEG; gold standard for seizure characterization and PNES diagnosis |
| Depth electrodes (SEEG) | Intracranial electrodes for pre-surgical epilepsy mapping |
| Electrocorticography (ECoG) | Intraoperative cortical surface recording |
| High-density EEG | 128-256 electrode arrays; better source localization |
| Quantitative EEG (qEEG) | Computer-processed spectral analysis; used in ICU monitoring |
| Magnetoencephalography (MEG) | Measures magnetic fields generated by the same currents; better spatial resolution, especially for deep sources |
10. EEG in Relation to Drug Effects
| Drug Class | EEG Effect |
|---|---|
| Benzodiazepines | Increased beta activity |
| Barbiturates | Increased beta; in withdrawal, burst suppression |
| Opioids | Decreased alpha; increased theta/delta voltage |
| Antipsychotics | Decreased alpha; increased theta |
| Alcohol (chronic) | Decreased alpha; during withdrawal, fast activity, photoparoxysmal responses |
| Hypothyroidism | Normal configuration but decreased amplitude and frequency |
- Kaplan & Sadock's Synopsis of Psychiatry
Summary: EEG records cortical electrical activity reflecting summated synaptic potentials, not individual action potentials. The 10-20 system standardizes electrode placement. Normal rhythms (alpha, beta, theta, delta) correspond to specific states of vigilance. Abnormal patterns - focal slowing, epileptiform discharges, triphasic waves, burst suppression, electrocerebral silence - each carry specific diagnostic implications. EEG remains irreplaceable in epilepsy classification, encephalopathy assessment, and coma evaluation, while newer applications in BCI continue to expand.
stages of sleep cycle
Reading File
Finding Sources
Searching the Web
stages of sleep cycle NREM REM physiology neuroscience 2024
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
I now have comprehensive content from multiple authoritative sources. Let me compile the full detailed answer.
Stages of the Sleep Cycle
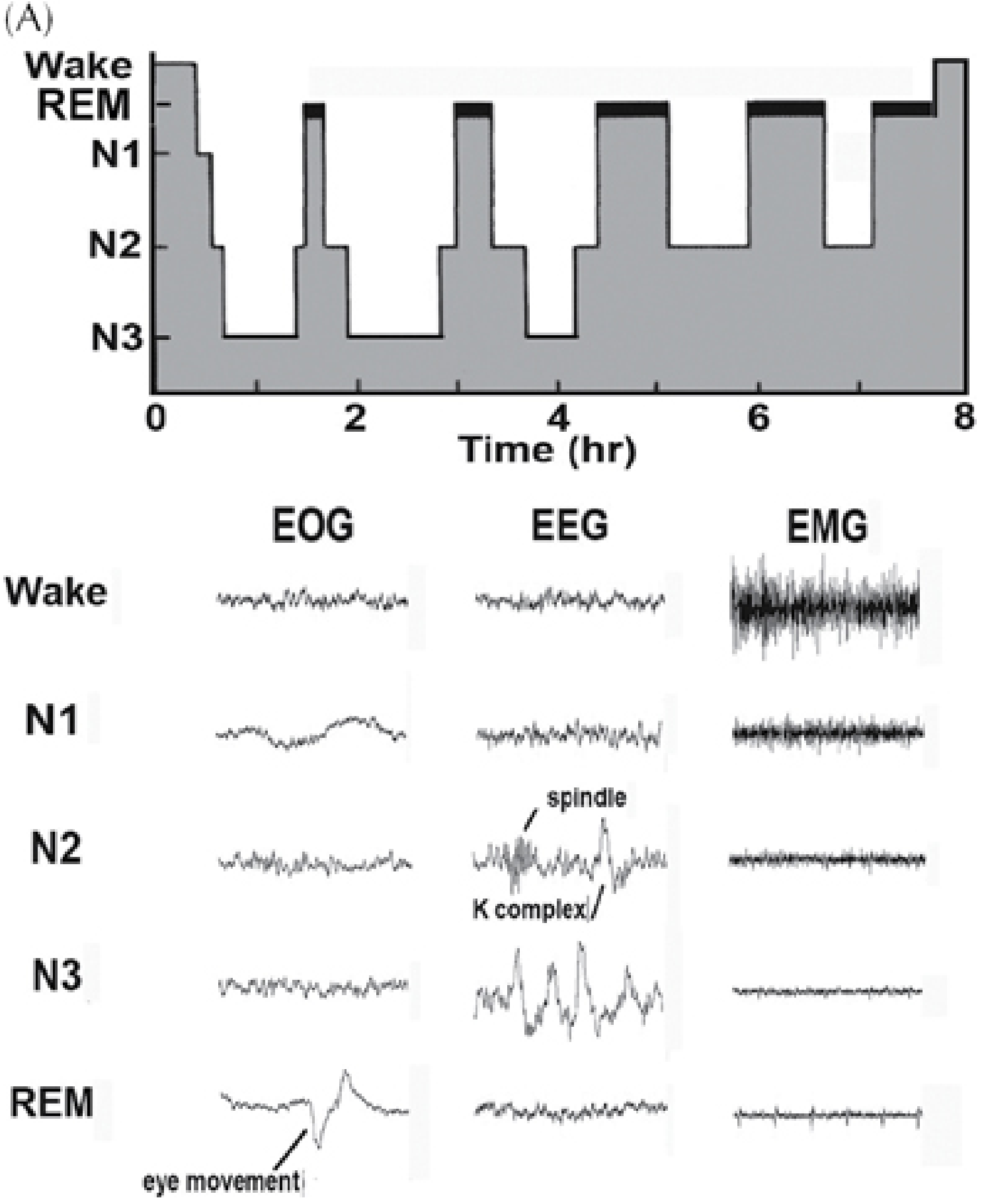
Figure: Sleep hypnogram (top) showing 4-5 NREM-REM cycles over 8 hours; bottom panel shows EOG, EEG, and EMG signatures for each stage. Note sleep spindle and K-complex in N2, high-voltage slow waves in N3, and REM atonia with eye movements - Plum and Posner's Diagnosis and Treatment of Stupor and Coma
1. Overview and Measurement
Sleep is not a single uniform state - it is an active, organized process with distinct stages that cycle throughout the night. The stages are defined by three simultaneous electrophysiologic recordings forming polysomnography (PSG):
- EEG (electroencephalogram) - brain wave patterns
- EOG (electro-oculogram) - eye movement activity
- EMG (electromyogram, surface chin/leg electrodes) - muscle tone
The current standard classification is from the American Academy of Sleep Medicine (AASM) and recognizes five stages:
| Stage | Category | Old Terminology |
|---|---|---|
| W | Wakefulness | Stage Wake |
| N1 | NREM sleep | Stage 1 |
| N2 | NREM sleep | Stage 2 |
| N3 | NREM slow-wave sleep | Stages 3 + 4 combined |
| R | REM sleep | Stage REM |
- Adams and Victor's Principles of Neurology, 12th Ed.
- Harrison's Principles of Internal Medicine, 22nd Ed.
2. Stage W - Wakefulness
EEG: Posterior alpha rhythm (8-12 Hz, ~50 µV) when eyes are closed and relaxed; replaced by low-voltage, irregular beta activity when eyes open or during mental effort.
EOG: Voluntary, spontaneous eye movements.
EMG: Active - muscle tone is highest here.
The transition from wakefulness to sleep is marked by:
- Eyes beginning to droop and slow, roving lateral eye movements appearing
- Pupils constricting
- Muscles relaxing progressively
- Alpha waves dropping out and replaced by low-voltage mixed-frequency activity
3. Stage N1 - Light NREM Sleep
Duration: 1-7 minutes per episode; comprises only 3-5% of total sleep time
EEG:
- Loss of alpha waves
- Low-voltage, mixed-frequency activity (predominantly theta, 4-7 Hz)
- Vertex sharp waves may appear at central electrode sites (Cz), especially in younger individuals
EOG: Slow, roving eye movements (no rapid movements)
EMG: Reduced but still present muscle tone
Subjective experience: This is the transitional zone - subjects may feel they have not been asleep. Many people hypnic jerks (sudden muscle contractions) during N1. Stage N1 is the lightest sleep; easy arousal with a modest stimulus.
Physiology:
- Blood pressure begins to fall slightly
- Body temperature begins to decrease
- Heart rate slows slightly
4. Stage N2 - Intermediate NREM Sleep
Duration: Longest single stage, comprising 50-60% of total sleep time
EEG - defining features (both must be present):
- Sleep spindles - bursts of 12-14 Hz waxing-and-waning activity lasting 0.5-2 seconds; maximal over biparietal/central regions; generated by thalamocortical circuits (thalamic reticular nucleus gating)
- K-complexes - high-amplitude, biphasic (negative sharp wave followed by slow positive deflection) complexes at central-parietal regions; can be triggered by external stimuli or occur spontaneously; thought to represent a cortical response suppressing arousal
EOG: Slow or absent eye movements; no rapid eye movements
EMG: Low, reduced further from N1
Physiology:
- Blood pressure and heart rate progressively lower
- Body temperature drops further
- Sleep bruxism (teeth grinding) and sleep talking can occur in N2
- Most sleepwalking actually originates from N2/N3 transitions
As the night progresses, N2 becomes the dominant stage in later cycles after N3 diminishes.
5. Stage N3 - Slow-Wave Sleep (Deep NREM / SWS)
Duration: 15-25% of total sleep time in young adults; predominantly in the first third of the night
EEG - defining features:
- Delta waves (0.5-2 Hz, >75 µV amplitude) comprising ≥20% of the EEG (some guidelines say ≥20% of a 30-second epoch)
- High-voltage, synchronized slow activity
- Sleep spindles and K-complexes disappear
EOG: Minimal or absent eye movements
EMG: Very low muscle tone
Behavioral characteristics:
- Hardest stage to arouse from - stimuli that would easily wake someone from N1 or N2 may fail here
- If aroused from N3, the person typically feels disoriented and groggy for several minutes ("sleep inertia")
- Parasomnias arising from N3: sleepwalking (somnambulism), sleep terrors, confusional arousals, and sleep-related eating disorder - the person is partially awake and partially in deep sleep
Physiological changes:
- Blood pressure falls 10-20% below daytime values
- Heart rate at its lowest
- Respiratory rate slow and regular
- Basal metabolic rate decreases ~10-30%
- Growth hormone (GH) - the largest secretory pulse of the night occurs in the first N3 episode; SWS is the primary driver of GH release
- Immune restoration - cytokine release and immune function consolidation
- This is the most restorative form of sleep; sleep deprivation causes selective N3 rebound on recovery nights
6. Stage R - REM Sleep (Rapid Eye Movement / Paradoxical Sleep)
First described in 1953 by Aserinsky and Kleitman, who observed that subjects periodically entered a state where their EEG resembled wakefulness, yet they were deeply unresponsive to external stimuli with their eyes closed.
Duration: 20-25% of total sleep time in young adults; REM periods become progressively longer through the night; the last REM episode (near morning) may last 30-45 minutes
EEG:
- Low-voltage, mixed-frequency, desynchronized - similar to wakefulness or N1
- Sawtooth waves (2-6 Hz notched waves) may appear, often preceding bursts of rapid eye movements
- Called "paradoxical sleep" because the EEG looks awake while the person is behaviorally asleep
EOG: Rapid, conjugate, often binocular eye movements occurring in bursts ("phasic REM")
EMG: Nearly silent - generalized skeletal muscle atonia (paralysis) mediated by the brainstem
Key characteristics of REM sleep:
- Active dreaming - complex, narrative, emotionally vivid dreams occur predominantly during REM; subjects awakened during REM recall dreams most consistently
- Muscle atonia - brainstem-mediated inhibition of spinal motor neurons; serves to prevent acting out dreams (failure = REM sleep behavior disorder)
- Autonomic instability - heart rate and respiration become irregular, unlike the regularity of NREM
- Penile/clitoral tumescence occurs in REM; used clinically to distinguish psychogenic from organic erectile dysfunction (nocturnal penile tumescence testing)
- Brain metabolism increases up to 20% above wakefulness levels in some areas
- Increased arousal threshold - paradoxically, despite active EEG, REM can be harder to arouse from than NREM
- Temperature dysregulation - poikilothermy (body temperature follows environmental temperature rather than being actively regulated)
Functions of REM sleep:
- Memory consolidation - particularly procedural and emotional memories; learning and synaptic plasticity
- Emotional processing - threat simulation theory; amygdala highly active
- Neural development - explains why newborns spend ~50% of sleep in REM (high plasticity period)
7. The Ultradian Sleep Cycle
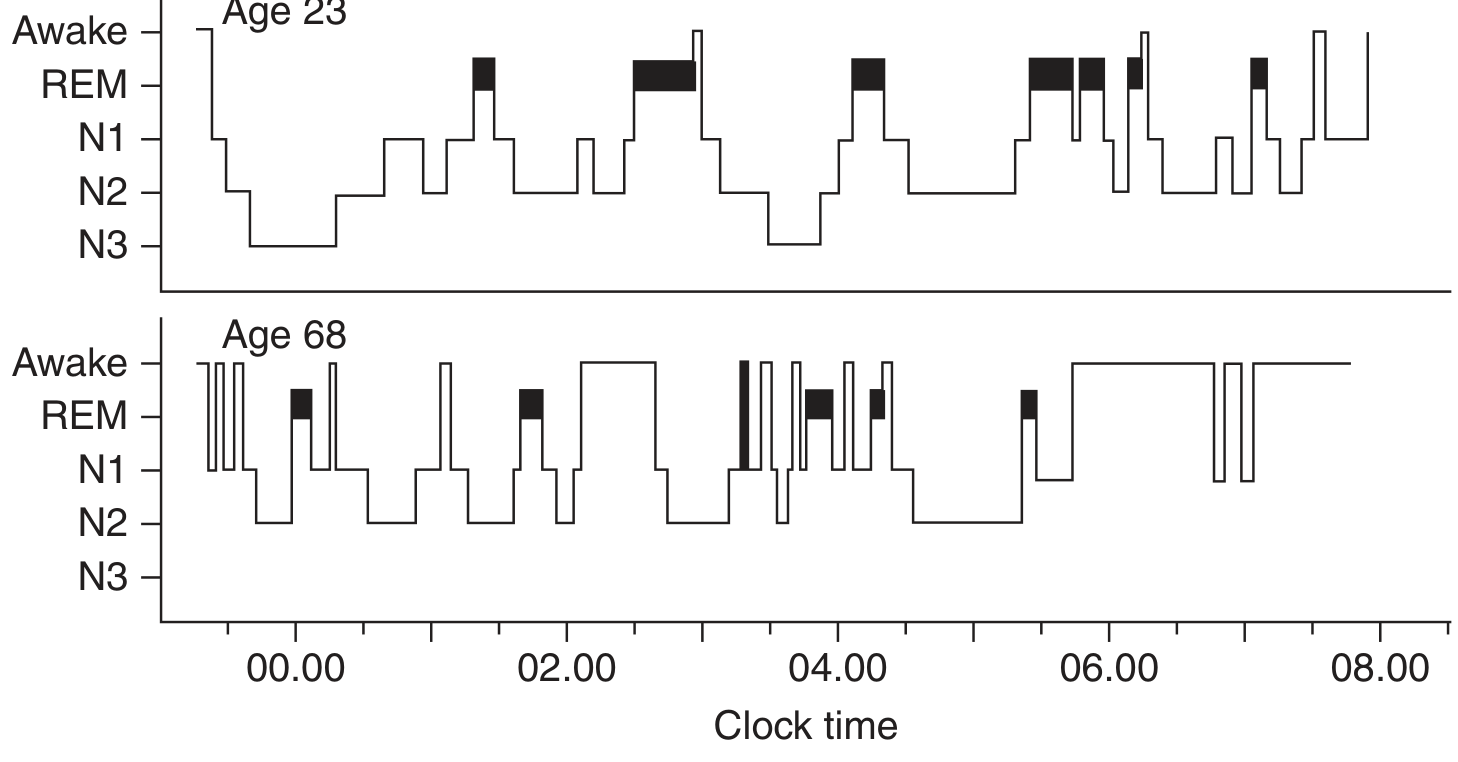
Figure: Hypnograms comparing sleep architecture at age 23 vs 68. Note rich N3 and well-organized REM cycles in the young adult, versus fragmented, shallow sleep with absent N3 in the older adult - Harrison's Principles of Internal Medicine, 22nd Ed.
A normal night's sleep follows this progression:
- Sleep onset → N1 → N2 → N3 (rapid descent in 45-60 min)
- First REM episode at ~90 minutes, usually brief (5-10 min)
- NREM-REM cycle repeats 4-6 times per night with ~90-100 minute periodicity ("ultradian rhythm")
- Early night is dominated by N3 (deep NREM)
- Late night is dominated by REM (REM episodes grow longer; N3 may be absent)
- Brief awakenings from lighter NREM or REM during the night are normal and usually not recalled
Proportions of sleep by stage (young adults):
| Stage | % of Total Sleep Time |
|---|---|
| N1 | 3-5% |
| N2 | 50-60% |
| N3 | 15-25% |
| REM | 20-25% |
8. Sleep Across the Lifespan
| Age Group | Key Features |
|---|---|
| Newborns | 50% REM; 60-minute cycle; enter REM directly at sleep onset ("active sleep") |
| Infants (3-6 mo) | Cycle lengthens; N3 increases; REM percentage falls |
| Children | Most intense N3 of lifespan; long sleep duration (9-12 hrs) |
| Adolescents | N3 begins to decline; delayed circadian phase ("night owl") |
| Young adults (18-25) | Peak sleep organization; normal proportions above |
| Middle age | N3 progressively decreases; more awakenings |
| Elderly (>65) | N3 may be virtually absent; frequent awakenings; earlier bedtime/wake; more fragmented sleep; REM preserved but reduced |
Sleep deprivation increases the rapidity of sleep onset and both the intensity and amount of slow-wave sleep on recovery nights, demonstrating homeostatic sleep pressure (Process S).
9. Neural Control of Sleep
Two processes govern timing of sleep:
Process S - Homeostatic Sleep Pressure
- Adenosine accumulates in the basal forebrain during wakefulness and acts as a sleep-promoting substance
- Adenosine levels rise proportionally with waking duration and fall during sleep
- Caffeine works by blocking adenosine receptors (A1 and A2A), thereby preventing the sleepiness signal
Process C - Circadian Timing
- The suprachiasmatic nucleus (SCN) of the anterior hypothalamus is the master pacemaker
- Entrains to light-dark cycle via the retinohypothalamic tract
- Controls nocturnal release of melatonin from the pineal gland (darkness triggers release; light suppresses it)
- Melatonin signals "biological night" and promotes sleep onset
The Flip-Flop Switch
Sleep-wake transitions are not gradual but sharp, governed by mutual inhibition:
Wake-promoting systems:
- Locus coeruleus (norepinephrine)
- Dorsal raphe (serotonin)
- Tuberomammillary nucleus (histamine) - explains why antihistamines cause drowsiness
- Pedunculopontine/laterodorsal tegmental nuclei (acetylcholine)
Sleep-promoting system:
- Ventrolateral preoptic nucleus (VLPO) of the hypothalamus - releases GABA and galanin to inhibit all wake-promoting centers
These systems mutually inhibit each other, creating a bistable switch that produces sharp transitions between wake and sleep. Orexin (hypocretin), released by lateral hypothalamic neurons, stabilizes the switch toward wakefulness - loss of orexin neurons causes narcolepsy, destabilizing both the wake/sleep and NREM/REM switches (resulting in cataplexy, sleep paralysis, and hypnagogic hallucinations).
REM Generation
REM sleep is generated by cholinergic neurons in the pontine tegmentum (pedunculopontine and laterodorsal tegmental nuclei). A separate "REM flip-flop" in the pons controls transitions between REM and NREM via GABAergic populations. During REM, monoaminergic (norepinephrine, serotonin) neurons are nearly silent, while cholinergic neurons are highly active.
- Adams and Victor's Principles of Neurology, 12th Ed.
- Guyton and Hall Textbook of Medical Physiology
- Harrison's Principles of Internal Medicine, 22nd Ed.
10. Physiological Summary Table
| Parameter | Wake | N1 | N2 | N3 | REM |
|---|---|---|---|---|---|
| EEG | Alpha/Beta | Mixed theta | Spindles + K-complexes | Delta (slow waves) | Low-voltage mixed (like wake) |
| Eye movements | Voluntary | Slow roving | Slow/absent | Absent | Rapid (bursts) |
| Muscle tone | High | Reduced | Low | Very low | Near absent (atonia) |
| Heart rate | Normal | Slightly reduced | Low | Lowest | Irregular |
| Respiration | Normal | Slowing | Regular, slow | Very regular, slow | Irregular |
| Temperature regulation | Active | Active | Active | Active | Poikilothermic |
| Growth hormone | Basal | - | - | Peak pulse | Low |
| Dreams | Rare | Occasional | Occasional | Rare (passive) | Vivid, narrative |
| Arousal ease | Easy | Easy | Moderate | Difficult | Paradoxically difficult |
chemical mediators of sleep
Finding Sources
Searching the Web
chemical mediators of sleep adenosine orexin GABA melatonin neuroscience 2024 2025
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Loading Image
Finding Sources
I now have rich, comprehensive content from multiple textbook sources with clear diagrams. Let me compile the full detailed answer.
Chemical Mediators of Sleep
The sleep-wake cycle is governed by two opposing forces - wake-promoting systems (ascending arousal system) and sleep-promoting systems - balanced through a "flip-flop switch" mechanism. Below is a systematic account of every major chemical mediator.
The Big Picture: Two Drives, One Switch
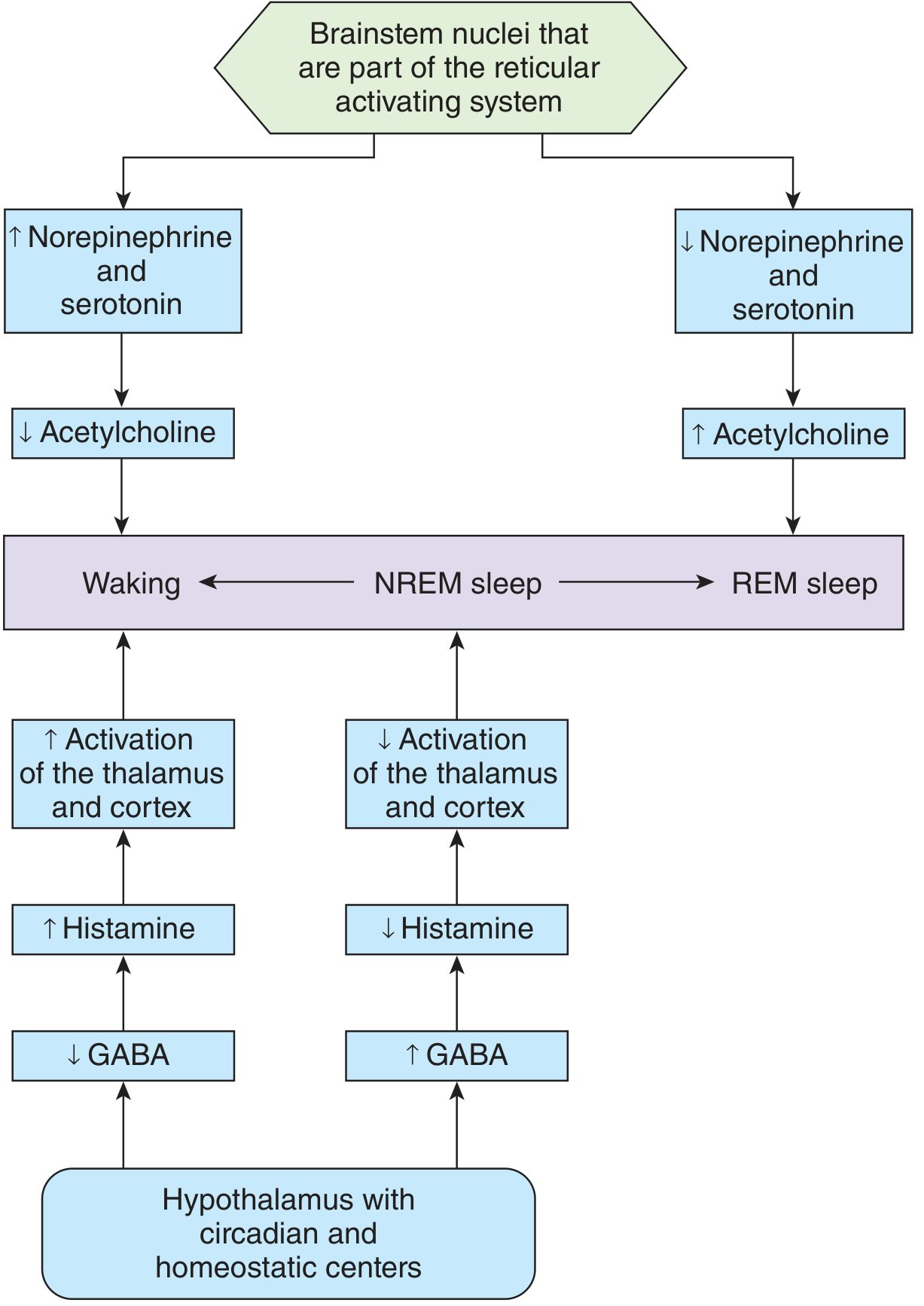
Figure: Model of how brainstem and hypothalamic neurotransmitters govern the three states - waking (high monoamines, low ACh), NREM (balanced), and REM sleep (low monoamines, high ACh). Histamine and GABA regulate thalamic/cortical activation. - Ganong's Review of Medical Physiology, 26th Ed.
Sleep timing is set by two interacting processes:
- Process S (Homeostatic): adenosine accumulates during wakefulness, building sleep pressure
- Process C (Circadian): the suprachiasmatic nucleus (SCN) drives a ~24-hour alternating wake/sleep signal modulated by light and melatonin
I. SLEEP-PROMOTING MEDIATORS
1. GABA (gamma-aminobutyric acid)
The primary inhibitory neurotransmitter of sleep.
Source: Neurons of the ventrolateral preoptic nucleus (VLPO) of the anterior hypothalamus - the master sleep-generating center.
Mechanism:
- During sleep, VLPO GABAergic neurons fire and release GABA + galanin onto all the major wake-promoting nuclei: the tuberomammillary nucleus (TMN), locus coeruleus (LC), dorsal raphe, ventral tegmental area (VTA), pedunculopontine/laterodorsal tegmental nuclei (PPT/LDT), and the basal forebrain
- This inhibition silences the arousal system and allows sleep to proceed
- GABA acts on GABA-A receptors (ionotropic Cl⁻ channels) → neuronal hyperpolarization → suppression of arousal
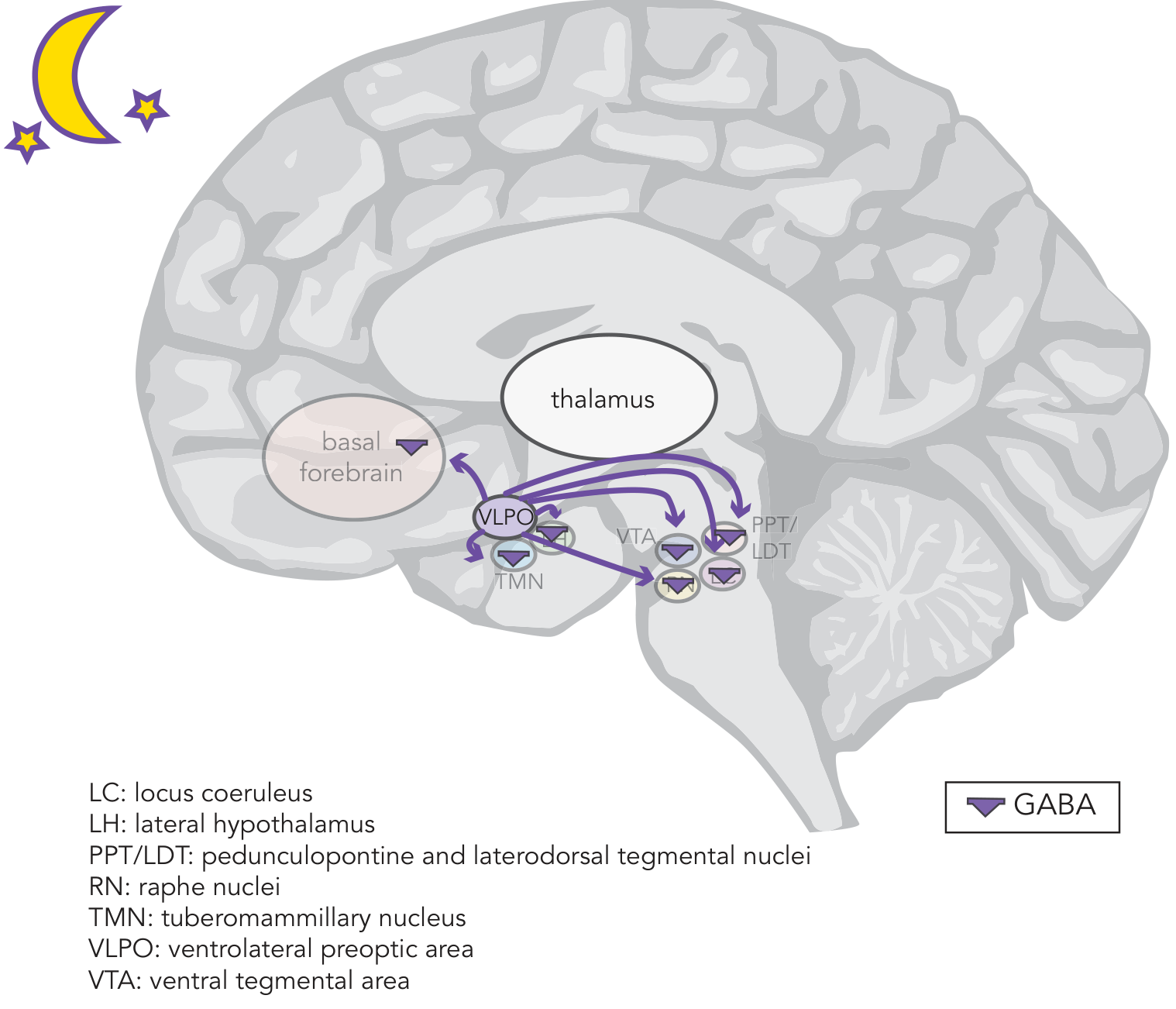
Figure: The sleep circuit. VLPO neurons release GABA (purple triangles) broadly to suppress the TMN, LC, raphe, VTA, PPT/LDT, and basal forebrain - silencing all wake-promoting pathways during sleep. - Stahl's Essential Psychopharmacology
Flip-flop switch: The VLPO and wake-promoting monoaminergic centers mutually inhibit each other. This creates a bistable switch: once tipped toward sleep, the system tends to remain asleep (VLPO inhibits monoamines, removing their inhibition of VLPO). The switch produces sharp transitions.
Pharmacology: Benzodiazepines and non-benzodiazepine hypnotics ("Z-drugs" - zolpidem, zaleplon, eszopiclone) act by potentiating GABA-A receptors, reducing sleep latency and increasing total sleep time. Barbiturates act similarly but with a wider therapeutic window.
- Stahl's Essential Psychopharmacology
- Eric Kandel - Principles of Neural Science, 6th Ed.
2. Galanin
Co-transmitter with GABA in VLPO neurons.
- Released alongside GABA from VLPO neurons during sleep
- Acts on galanin receptors (GalR1, GalR2) - inhibitory Gi-coupled GPCRs
- Reinforces the inhibitory silencing of TMN histaminergic neurons
- Also promotes REM sleep suppression mechanisms
3. Adenosine
The primary homeostatic sleep pressure signal.
Source: Produced as a metabolic byproduct of neuronal activity; accumulates in the extracellular space of the basal forebrain and other regions during sustained wakefulness. Glial cells also contribute to adenosine release.
Mechanism:
- Adenosine levels rise proportionally with duration of wakefulness
- Acts on A1 receptors on wake-promoting neurons → direct inhibition (hyperpolarization)
- Acts on A2A receptors on neurons in the nucleus accumbens shell and other regions → indirect disinhibition of VLPO, allowing sleep-promoting circuits to activate
- The resulting VLPO disinhibition promotes GABA release, which then silences arousal systems
- Adenosine levels fall during sleep as they are cleared - representing restoration of homeostatic sleep pressure
Caffeine mechanism: Adenosine A1 and A2A receptor antagonist. By blocking adenosine receptors, caffeine prevents the sleepiness signal from being perceived, maintaining wakefulness despite adenosine accumulation. This explains why caffeine delays but does not eliminate the sleep drive - when caffeine wears off, pent-up adenosine is still present and causes a "crash."
Sleep deprivation rebound: When sleep-deprived subjects finally sleep, their high adenosine levels drive increased N3 (slow-wave) sleep - the brain "catches up" on homeostatic sleep debt.
- Stahl's Essential Psychopharmacology
- Ganong's Review of Medical Physiology, 26th Ed.
4. Melatonin
The hormonal signal of biological night.
Source: Pineal gland; pathway: Tryptophan → 5-HTP → Serotonin → N-acetylserotonin (via NAT) → Melatonin (via HIOMT, hydroxyindole-O-methyltransferase)
Regulation:
- Secretion is triggered by darkness and suppressed by light (especially blue-spectrum, 480 nm)
- Light input travels via the retinohypothalamic tract → SCN → superior cervical ganglion (sympathetic) → pineal gland
- In humans, melatonin rises sharply ~2 hours before habitual bedtime ("dim-light melatonin onset", DLMO), peaks in the middle of the night, and falls before morning awakening
Mechanism:
- Acts on MT1 and MT2 receptors (Gi-coupled GPCRs) in the SCN and other brain regions
- MT1 activation: inhibits SCN neuronal firing → reduces the wake-promoting circadian signal
- MT2 activation: phase-shifts the circadian clock, mediating the entraining effects of light/dark cycles
- Does not directly cause sleep but lowers the threshold for sleep onset by suppressing circadian wakefulness promotion
Clinical relevance:
- Exogenous melatonin is used for jet lag, shift-work disorder, and delayed sleep phase syndrome
- Ramelteon (MT1/MT2 agonist) is an FDA-approved hypnotic that targets this system
- Tasimelteon used for non-24-hour sleep-wake disorder in blind individuals
5. Melanin-Concentrating Hormone (MCH)
- Produced by neurons in the lateral hypothalamus and zona incerta
- Inhibitory neuropeptide that promotes sleep, particularly REM sleep
- MCH neurons are active during sleep and nearly silent during wakefulness
- Work alongside VLPO to suppress arousal; MCH neuron ablation reduces REM sleep in animal models
6. Prostaglandin D₂ (PGD₂)
- A prostaglandin produced in the brain, particularly in the subarachnoid space overlying the basal forebrain
- Levels rise during prolonged wakefulness and fever
- Acts on DP1 receptors on leptomeningeal cells, triggering adenosine release → secondary sleep promotion
- Explains the somnolence associated with infection/inflammation; also the mechanism by which aspirin/NSAIDs (PG synthesis inhibitors) can mildly impair sleep in some individuals
7. Cytokines (IL-1β and TNF-α)
- Interleukin-1β and tumor necrosis factor-α are somnogenic - they promote NREM/slow-wave sleep
- Released during immune activation (explaining sickness-induced sleepiness)
- Levels show circadian variation, peaking during sleep onset
- Promote N3 (slow-wave) sleep via direct effects on the VLPO and adjacent circuits
- Part of the brain's mechanism linking immune status to sleep-wake behavior
II. WAKE-PROMOTING MEDIATORS
8. Orexin / Hypocretin
The master stabilizer of wakefulness.
Source: ~10,000-80,000 neurons exclusively in the lateral hypothalamic area, perifornical area, and posterior hypothalamus.
Two peptides cleaved from a single precursor (prepro-orexin):
- Orexin A (33 amino acids) - binds both OX1R and OX2R
- Orexin B (28 amino acids) - binds selectively to OX2R
Two receptors:
- OX1R: coupled to intracellular Ca²⁺ increase + Na⁺/Ca²⁺ exchanger activation
- OX2R: increases NMDA glutamate receptor expression + inactivates GIRK channels
Mechanism:
Orexin neurons project widely throughout the brain. During the day (especially with activity, stress, hunger), orexin is released and excites all wake-promoting centers simultaneously:
- Stimulates acetylcholine release from basal forebrain (→ cortical arousal) and PPT/LDT (→ thalamic activation)
- Drives dopamine release from VTA (→ motivation, reward, wakefulness)
- Promotes norepinephrine release from locus coeruleus (→ arousal, attention)
- Increases serotonin release from raphe nuclei (→ wakefulness)
- Increases histamine release from TMN (→ cortical/thalamic arousal)
- Together these create robust, stable wakefulness
Negative feedback: As norepinephrine and serotonin accumulate during extended wakefulness, they feed back to inhibit orexin neurons in the lateral hypothalamus. With orexin withdrawn, the VLPO-GABA system takes charge and sleep follows.
Narcolepsy: Loss of orexin-producing neurons (autoimmune destruction, likely triggered by infection/HLA-DQB1*06:02 susceptibility) causes narcolepsy type 1. The flip-flop switch becomes unstable in both directions:
- Fragmented, unstable wakefulness → excessive daytime sleepiness
- Intrusion of REM components into wakefulness:
- Cataplexy (sudden muscle atonia triggered by strong emotion)
- Sleep paralysis (REM atonia persisting into awakening)
- Hypnagogic/hypnopompic hallucinations (dream imagery at sleep onset/offset)
- CSF orexin A levels <110 pg/mL are diagnostic
New pharmacology: Suvorexant and lemborexant are dual orexin receptor antagonists (DORAs) approved as hypnotics - they block OX1R + OX2R, reducing wakefulness drive to facilitate sleep onset.
- Stahl's Essential Psychopharmacology
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry
9. Histamine
The wake-maintenance signal of the hypothalamus.
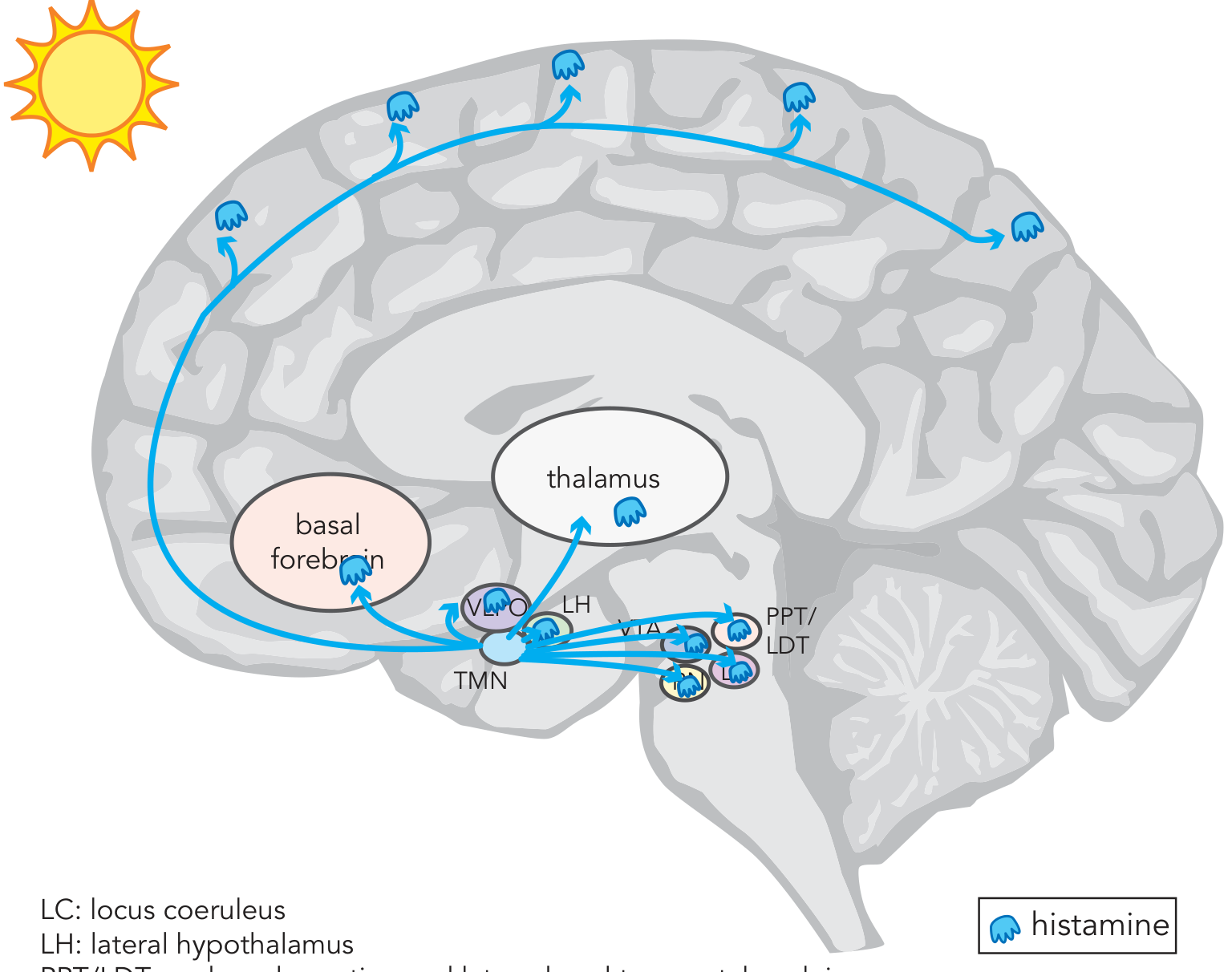
Figure: Histaminergic projections from the TMN to the prefrontal cortex, basal forebrain, thalamus, and brainstem centers. Histamine is the CNS's wakefulness maintenance transmitter. - Stahl's Essential Psychopharmacology
Source: Neurons of the tuberomammillary nucleus (TMN) of the posterior hypothalamus - the only brain histamine source.
Mechanism:
- TMN neurons are maximally active during wakefulness, slow during NREM, and nearly silent during REM
- Histamine projects to the prefrontal cortex, basal forebrain, thalamus, and all brainstem arousal centers
- Acts on H1 receptors (Gq) → depolarization of thalamic and cortical neurons → maintained wakefulness
- H1 antagonism (first-generation antihistamines: diphenhydramine, doxylamine) causes marked sedation - explaining their use as OTC sleep aids
- H3 receptors are autoreceptors on TMN neurons: H3 agonism reduces histamine release (used in treatment of narcolepsy/hypersomnia, e.g., pitolisant is an H3 inverse agonist)
GABA-histamine axis: Increased GABA from VLPO during sleep directly inhibits TMN neurons → histamine drops → thalamus and cortex deactivate → NREM sleep is maintained.
10. Norepinephrine (Noradrenaline)
Source: Locus coeruleus (LC) in the dorsal pons - the brain's main noradrenergic nucleus; projects to the entire neocortex, thalamus, hippocampus, cerebellum, and spinal cord.
State-dependence:
- LC neurons fire at highest rates during active wakefulness (especially during novelty, stress, attention)
- Firing decreases during quiet wakefulness and NREM sleep
- Nearly completely silent during REM sleep - a defining feature
Mechanism:
- Activates α1-adrenergic receptors (excitatory) and β-receptors on cortical neurons → promotes arousal and cognitive alertness
- Inhibits VLPO neurons via α2-adrenergic autoreceptors (negative feedback)
- Drives orexin release during active states
Pharmacology:
- Atomoxetine (NE reuptake inhibitor): promotes wakefulness; used in narcolepsy
- Clonidine (α2 agonist): reduces LC firing → sedation, used for ADHD/anxiety
- Antidepressants (TCAs, SNRIs) that increase NE → suppress REM sleep (explaining reduced REM% in patients on these drugs)
11. Serotonin (5-HT)
Source: Dorsal raphe nucleus (DRN) and median raphe nucleus; widespread projections to cortex, limbic system, thalamus, basal ganglia.
State-dependence:
- Most active during wakefulness
- Reduced during NREM
- Nearly silent during REM sleep (like LC)
Mechanism:
- Serotonin promotes wakefulness via 5-HT2A receptors on cortical neurons
- Simultaneously inhibits REM-generating cholinergic neurons in the PPT/LDT
- Activates wake-promoting basal forebrain cholinergic neurons (different population from REM-ACh)
- Orexin drives serotonin release to maintain daytime wakefulness
Dual role note: Some serotonin pathways act through different receptor subtypes to promote drowsiness/NREM (5-HT2A antagonism promotes deep sleep - the mechanism of some antipsychotics and mirtazapine that improve sleep quality).
Pharmacology:
- SSRIs and SNRIs increase serotonin → suppress REM sleep, reduce REM% (causing REM rebound on discontinuation)
- Trazodone (5-HT2A antagonist/SRI): promotes sleep, widely used as a hypnotic at low doses
- Mirtazapine (5-HT2A/5-HT3 + H1 antagonist): promotes deep NREM sleep, increases appetite
12. Dopamine
Source: Ventral tegmental area (VTA) and substantia nigra; projects to prefrontal cortex (mesocortical), limbic system (mesolimbic), and basal ganglia (nigrostriatal).
Role: Dopamine promotes wakefulness and motivated behavior but is less directly tied to sleep stage transitions than NE or serotonin. VTA neurons show highest activity during reward-related wakefulness.
- Dopamine reuptake transporter (DAT) blockade by modafinil and armodafinil is one mechanism behind their wake-promoting effects (though their full mechanism includes multiple targets)
- Amphetamines promote wakefulness partly via massive dopamine (and NE) release
- Dopamine D2 agonists (used in Parkinson's) can cause sudden sleep attacks
13. Acetylcholine (ACh)
The REM sleep generator and cortical arousal mediator.
Sources:
- Pedunculopontine nucleus (PPT) and laterodorsal tegmental nucleus (LDT) - pontine cholinergic cells - primarily drive REM sleep
- Basal forebrain cholinergic neurons (nucleus basalis of Meynert, medial septal nucleus) - primarily drive cortical arousal (wakefulness and REM)
State-dependence: Cholinergic neurons are active in two states: wakefulness AND REM sleep - both states share a desynchronized (low-voltage, fast) EEG. They are least active during NREM.
Mechanism in REM sleep:
- As monoaminergic (NE + serotonin) firing ceases at the NREM-REM transition, the cholinergic "REM-on" neurons in PPT/LDT become disinhibited
- ACh is released in the pontine reticular formation → generates the pontine-geniculate-occipital (PGO) waves that precede and accompany REM
- ACh from PPT/LDT activates thalamic relay neurons → desynchronized EEG (paradoxical wakefulness pattern)
- Descending ACh projections activate the REM atonia circuit via glutamatergic neurons in the sublaterodorsal area → inhibitory interneurons in the medulla/spinal cord hyperpolarize motor neurons
Pharmacology:
- Cholinergic agonists (physostigmine, pilocarpine): promote REM sleep; can trigger REM-onset nightmares
- Cholinergic antagonists (scopolamine, atropine): suppress REM sleep
- REM sleep behavior disorder (RBD): failure of REM atonia (loss of sublaterodorsal neurons) → patients physically act out vivid dreams; early marker of Lewy body disease/Parkinson's
III. SUMMARY TABLE
| Mediator | Source | Role | State Activity |
|---|---|---|---|
| GABA | VLPO | Core sleep inducer; inhibits all wake centers | Sleep |
| Galanin | VLPO | Co-inhibitor with GABA | Sleep |
| Adenosine | Neuronal/glial metabolism | Homeostatic sleep pressure; accumulates during wake | Rises during wake, clears during sleep |
| Melatonin | Pineal gland | Circadian "dark" signal; lowers sleep threshold | Night |
| MCH | Lateral hypothalamus | REM-promoting, sleep facilitation | Sleep/REM |
| PGD₂ | Subarachnoid space | Somnogen via adenosine release | Wake/sickness |
| IL-1β, TNF-α | Immune/glial cells | N3 promoters during illness | Inflammation |
| Orexin/Hypocretin | Lateral hypothalamus | Master wakefulness stabilizer; drives all arousal transmitters | Active wake |
| Histamine | TMN | Sustained cortical/thalamic arousal | Wake |
| Norepinephrine | Locus coeruleus | Arousal, attention, inhibits REM | Wake/NREM; silent in REM |
| Serotonin | Dorsal raphe | Wakefulness; REM suppression | Wake/NREM; silent in REM |
| Dopamine | VTA | Motivated wakefulness, arousal | Wake |
| Acetylcholine | PPT/LDT + Basal forebrain | REM generation; cortical desynchronization | Wake + REM |
IV. The Homeostatic-Circadian Interaction
As the day progresses:
- Adenosine accumulates in the basal forebrain → progressive disinhibition of VLPO
- The SCN's circadian wake-promoting signal (partly orexin-driven) counteracts adenosine during the day - explaining afternoon alertness despite hours awake
- In the early evening, SCN wake-promotion wanes, and unmasked adenosine tips the balance toward sleep
- Melatonin is released ~2 hours before bedtime, suppressing SCN firing and facilitating the transition
- VLPO fires → GABA floods all wake centers → sleep begins
- During sleep, adenosine clears → homeostatic debt repaid
- Near morning, circadian promotion of sleep occurs (prevents premature awakening), then light suppresses melatonin and SCN ramps up wakefulness signals → awakening
Caffeine's "debt" mechanism: Caffeine blocks adenosine receptors but does not stop adenosine accumulation. When it wears off (~5-6 hr half-life), the accumulated adenosine floods previously blocked receptors simultaneously, producing a pronounced "crash."
- Stahl's Essential Psychopharmacology
- Ganong's Review of Medical Physiology, 26th Ed.
- Eric Kandel - Principles of Neural Science, 6th Ed.
- Adams and Victor's Principles of Neurology, 12th Ed.
explain in detail about the reticular activating system use guyton and hall and ganong as your reference
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
The answer above is drawn exclusively from Guyton and Hall and Ganong as requested. Here is a concise roadmap of what was covered:
Quick Reference Index
| Section | Key Source |
|---|---|
| Anatomy: excitatory + inhibitory reticular areas | Guyton Ch. 59, Fig. 59.1 |
| Two signal types to cortex (fast ACh + slow sustained) | Guyton Ch. 59 |
| Sensory inputs driving the RAS; CN V transection experiment | Guyton Ch. 59 |
| Cortical positive feedback loop | Guyton Ch. 59 |
| Thalamus as gateway + two thalamic projection systems | Ganong Ch. 14 |
| ARAS four-component neurochemical model (LC, raphe, PPT/LDT, TMN) | Ganong Fig. 14-3 |
| Neurohormonal nuclei: NE, dopamine, serotonin, ACh in human brainstem | Guyton Fig. 59.3 |
| Wake/NREM/REM transitions: monoamine-ACh reciprocal model | Ganong Fig. 14-4 |
| Sleep as active inhibition (not RAS fatigue); midpontine transection | Guyton Ch. 60 |
| Orexin stabilization of RAS; narcolepsy | Guyton Ch. 60 |
| Sleep-inducing substances (muramyl peptide, delta peptide) | Guyton Ch. 60 |
| Clinical: coma, narcolepsy, insomnia from RAS lesions | Both |
This is a shared conversation. Sign in to Orris to start your own chat.