Essay on HLH
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hemophagocytic lymphohistiocytosis"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Searching Images
hemophagocytic lymphohistiocytosis bone marrow hemophagocytosis pathology
Reading File
I now have all the material needed for a comprehensive essay. Let me compose it.
Hemophagocytic Lymphohistiocytosis (HLH)
Definition and Overview
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening syndrome of pathologic immune hyperactivation — a "cytokine storm" — characterized by uncontrolled activation of CD8⁺ cytotoxic T lymphocytes and macrophages that leads to progressive organ failure. The term hemophagocytosis (also called erythrophagocytosis) describes the hallmark finding of macrophages engulfing red cells, platelets, and nucleated marrow cells in the bone marrow, spleen, and lymph nodes. Despite this dramatic histologic appearance, hemophagocytosis is neither pathognomonic nor invariably present at diagnosis; the syndrome is defined by a constellation of clinical, laboratory, and genetic findings rather than by morphology alone.
HLH exists in two broad forms: primary (genetic/familial) HLH, predominantly a disease of infancy and childhood caused by Mendelian defects in cytotoxic lymphocyte function, and secondary (acquired) HLH, more common in adults and triggered by infection, malignancy, or autoimmune disease. Despite entirely different root causes, both forms converge on an identical clinical phenotype — the convergent phenotype — of sepsis-like systemic inflammation progressing to multiorgan failure and, if untreated, death.
Epidemiology
Familial HLH (FHL) occurs in approximately 1 in 50,000 live births, though estimates range from 1 to >200 per 300,000 births depending on geography and ascertainment. Together with severe combined immunodeficiency, it is one of the most common fatal inherited immunodeficiencies. Consanguinity raises incidence, and no sex predilection exists. The median age of onset is 3–6 months, though very early (congenital) or late (adolescent/adult) presentations occur.
Secondary HLH in adults has been recognized as a distinct entity only in the past 15–20 years. Population-based studies report annual incidences of approximately 4.2 per million (England) and 6.2 per million for malignancy-associated HLH (Sweden). HLH appears more common in East Asia than in Western populations; EBV-HLH, in particular, disproportionately affects individuals of East Asian origin. — Harrison's Principles of Internal Medicine, 22E
Pathophysiology
The Central Defect: Failure of Cytotoxic Lymphocyte Termination
Under normal conditions, CD8⁺ cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells recognize and kill antigen-bearing target cells by releasing perforin-containing cytolytic granules. Perforin polymerizes to form pores in the target cell membrane, facilitating granzyme entry and apoptosis. Crucially, target cell elimination leads to antigen clearance, which downregulates the immune response — a process termed activation-induced cell death.
In primary HLH, homozygous null mutations in genes encoding proteins required for the synthesis, docking, and exocytosis of cytolytic granules abolish this kill-and-terminate mechanism. Unable to efficiently destroy their targets, CTLs remain in prolonged contact with antigen-presenting cells and secrete escalating amounts of inflammatory cytokines. The result is a vicious cycle: persistent antigen → persistent CTL activation → hypercytokinemia → macrophage activation → further cytokine release → tissue destruction.
Cytokine Storm
The dominant cytokine driving HLH pathology is interferon-γ (IFN-γ), released in massive quantities by overactivated CTLs and NK cells. IFN-γ activates macrophages and induces expression of CXCL9, a chemokine used as a biomarker of IFN-γ signaling. Additional inflammatory mediators include IL-1β, IL-6, IL-18, and TNF-α. A key diagnostic distinction from sepsis: in HLH, IFN-γ is markedly elevated with moderate IL-6, whereas in bacterial sepsis the reverse pattern (high IL-6, moderate IFN-γ) predominates. — Harrison's 22E; Robbins & Kumar Basic Pathology
Why Macrophages Phagocytose Blood Cells
Macrophages activated by IFN-γ and other cytokines upregulate phagocytic activity indiscriminately, engulfing red cells, platelets, and leukocyte precursors. This hemophagocytosis directly contributes to cytopenias and provides the eponymous pathologic appearance.
Classification
Table: Simplified Classification of HLH (Harrison's 22E, Table 68-1)
| Category | Subtypes |
|---|---|
| Primary HLH (Mendelian) | FHL2 (PRF1), FHL3 (UNC13D), FHL4 (STX11), FHL5 (STXBP2); XLP1 (SH2D1A); Griscelli syndrome type 2 (RAB27A); Chédiak-Higashi; XLP2 (BIRC4/XIAP); NLRC4 inflammasomopathy; lysinuric protein intolerance; Wolman disease |
| Secondary HLH (non-Mendelian) | Infection-associated (viral, bacterial, parasitic, fungal); Malignancy-associated; Autoimmune-associated (MAS-HLH: SoJIA, AOSD, SLE, vasculitis); Transplant-associated |
Primary (Familial) HLH
Genetics
FHL follows autosomal recessive inheritance and is caused by biallelic loss-of-function variants in four core genes: PRF1 (perforin; ~30%), UNC13D (Munc13-4; ~30%), STXBP2 (Munc18-2; ~20%), and STX11 (syntaxin-11; ~10%). Each of these proteins plays a distinct role in granule priming, docking, or membrane fusion. Variant frequency differs by ethnicity; specific founder mutations are found in certain populations (e.g., PRF1 p.A91V in European populations, splice-site variants in UNC13D in Korean patients).
HLH with Partial Albinism (Immune + Pigmentation Syndromes)
Three conditions combine HLH with abnormal pigmentation, and hair examination can assist diagnosis:
- Griscelli syndrome type 2 (RAB27A mutation): silver-gray hair, immune defects
- Chédiak-Higashi syndrome (LYST mutation): silver hair, giant cytoplasmic granules in leukocytes on blood smear, progressive neurological disease
- Hermansky-Pudlak syndrome type II (AP3B1 mutation): oculocutaneous albinism
X-linked Lymphoproliferative Syndrome
XLP1 (SH2D1A/SAP deficiency) typically presents in preschool-aged males with HLH precipitated by EBV infection. SAP normally facilitates 2B4-mediated NK cytotoxicity and T-cell co-stimulation; its absence results in inability to control EBV-infected B cells. One-third develop lymphoma; allogeneic HSCT should be considered early. Rituximab (anti-CD20) targets the B-cell reservoir of EBV and is added to HLH-directed therapy.
XLP2 (XIAP/BIRC4 deficiency) presents similarly but is often milder and is frequently associated with Crohn's-like inflammatory bowel disease. Myeloablative conditioning for HSCT carries higher risk in this subtype.
NLRC4 Inflammasomopathy
Gain-of-function variants in NLRC4 (part of the inflammasome) cause severe early-onset HLH with markedly elevated serum IL-18. Targeted IL-18 inhibition is effective therapy for this subtype.
Secondary HLH
In adults with secondary HLH, the three major trigger categories and their approximate frequencies are: malignancy (~48%), infection (~50%), and autoimmune disease (~13%), with overlap common.
Infection-Associated HLH
Viral infections account for ~70% of infection-associated cases. Herpesviruses dominate: EBV causes ~40% of viral cases (the single most common trigger overall), CMV ~10%. Other notable viral triggers include hepatitis viruses, parvovirus B19, influenza, HIV, dengue (HLH found in ~10% of severe dengue, 50% mortality), and SARS-CoV-2. Bacterial triggers include M. tuberculosis, Rickettsia spp., Staphylococcus, and E. coli. Parasitic HLH is most often caused by visceral leishmaniasis and Plasmodium; fungal HLH by Histoplasma spp.
Malignancy-Associated HLH
Lymphoma (particularly T-cell and NK-cell lymphomas) is the trigger in >50% of adult HLH. B-cell lymphomas and angioimmunoblastic T-cell lymphoma are also implicated. In malignancy-associated HLH, aberrant cytokine production by neoplastic lymphocytes dysregulates the immune response. Treatment of the underlying malignancy — usually with an etoposide-containing regimen — should be initiated immediately alongside HLH-directed immunosuppression.
Macrophage Activation Syndrome (MAS-HLH)
HLH in the context of autoimmune disease is termed macrophage activation syndrome (MAS). It most commonly complicates systemic-onset juvenile idiopathic arthritis (SoJIA) and adult-onset Still's disease (AOSD), as well as systemic lupus erythematosus and vasculitis. A significant proportion of MAS patients carry heterozygous variants in FHL-associated genes, creating a genetic "second hit" susceptibility. MAS may be mistaken for a disease flare; a disproportionate rise in ferritin, falling ESR (paradoxical since fibrinogen is consumed), thrombocytopenia, and rising transaminases should raise suspicion.
Clinical Features
The typical presentation is a sepsis-like picture that does not respond to antimicrobial therapy:
| System | Manifestations |
|---|---|
| Constitutional | Prolonged fever (often >38.5°C, spiking pattern) |
| Reticuloendothelial | Hepatosplenomegaly (almost universal); lymphadenopathy (~50%) |
| Hematologic | Bicytopenia or pancytopenia; thrombocytopenia is earliest and most severe; petechiae, purpura |
| Hepatic | Elevated transaminases, conjugated hyperbilirubinemia (jaundice), elevated γ-GT; may mimic acute liver failure |
| Coagulation | DIC; low fibrinogen; bleeding |
| Metabolic | Hypertriglyceridemia; hyponatremia; hypoalbuminemia |
| Neurologic | Seizures, altered consciousness, meningism, cranial nerve palsies, ataxia (more common in children; ~⅓ of FHL patients at diagnosis) |
| Dermatologic | Transient maculopapular rash; edema |
Ferritin is almost universally elevated and may reach strikingly high values (>10,000 μg/L is strongly suggestive). Soluble IL-2 receptor (sIL-2r / sCD25) is another sensitive inflammatory marker reflecting T-cell activation.
In adults, CNS involvement is less common than in children. Adults with secondary HLH often present with critical illness, multiorgan failure, and an undiagnosed malignancy.
Diagnosis
HLH-2004 Criteria
Diagnosis requires either (1) molecular confirmation of a familial HLH gene variant, or (2) fulfillment of ≥5 of 8 clinical/laboratory criteria:
- Fever
- Splenomegaly
- Bicytopenia (≥2 cytopenias: Hb <90 g/L, platelets <100 × 10⁹/L, neutrophils <1.0 × 10⁹/L)
- Hypertriglyceridemia (fasting TG ≥3.0 mmol/L / ≥265 mg/dL) and/or hypofibrinogenemia (≤1.5 g/L)
- Ferritin ≥500 μg/L
- Soluble CD25 (sIL-2r) ≥2400 U/mL
- Hemophagocytosis in bone marrow, spleen, or lymph nodes
- Low or absent NK-cell activity
Note: The Histiocyte Society revised these criteria in 2024; the sCD25 threshold and other parameters are under review.
HScore
The HScore is an alternative scoring tool designed for adults that incorporates 9 variables (temperature, organomegaly, cytopenias, ferritin, triglycerides, fibrinogen, AST, immunosuppression status, and known underlying immunosuppressive disease) to estimate the probability of HLH. An HScore >169 carries ~93% specificity; it performs well alongside HLH-2004 criteria in critically ill adults.
Functional and Genetic Testing
- NK-cell cytotoxicity assay (flow cytometry): impaired in FHL and related disorders
- CD107a degranulation assay: measures perforin granule exocytosis — most practical first-line functional screen
- Intracellular perforin staining: rapid screen for PRF1-related disease
- SAP/XIAP expression: screens for XLP1/XLP2
- Next-generation sequencing gene panel: gold standard for confirming primary HLH, but results may take weeks
Workup for Underlying Triggers
Every patient requires a thorough evaluation: blood cultures; viral PCR/serologies (EBV, CMV, HIV, HHV-6/8, influenza, parvovirus B19, hepatitis B/C, HSV, VZV, adenovirus); autoimmune panel; and — critically — staging for lymphoma (CT chest/abdomen/pelvis or PET-CT; bone marrow biopsy; TCR gene rearrangement studies). Because lymphoma is the trigger in ~50% of adult HLH, missing it is fatal.
Pathology
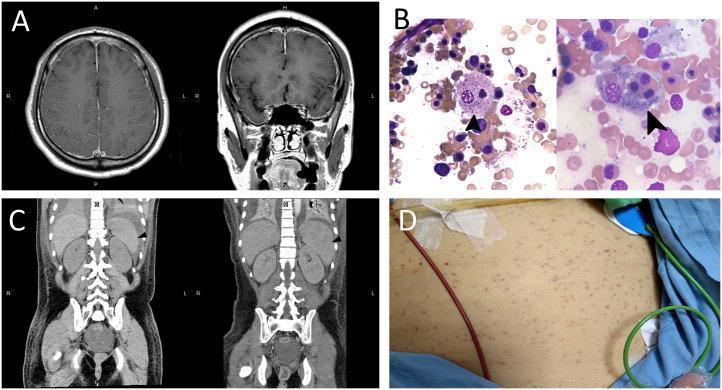
Bone marrow biopsy or aspirate demonstrates the hallmark finding: activated macrophages (histiocytes) engulfing erythrocytes, platelets, and nucleated hematopoietic cells (hemophagocytosis). This finding is not specific to HLH — it can be seen in sepsis, multiple transfusions, and other inflammatory states — and may be absent in early disease. Serial biopsies may be needed. The marrow may become progressively hypocellular.
Treatment
General Principles
Treatment rests on two pillars: (1) identify and treat the underlying trigger and (2) suppress the hyperinflammatory response with HLH-directed therapy proportional to disease severity. Early diagnosis is critical — delay results in CNS damage, irreversible organ failure, and death.
HLH-94 and HLH-2004 Protocols (Primary HLH / Severe Secondary HLH)
The international HLH-94 protocol, validated by the Histiocyte Society, remains the foundation of FHL treatment:
Initial 8-week induction:
- Dexamethasone: 10 mg/m²/day (weeks 1–2) → 5 mg/m²/day (weeks 3–4) → 2.5 mg/m²/day (weeks 5–6) → 1.25 mg/m²/day (week 7) → taper week 8
- Etoposide: 150 mg/m² IV twice weekly (weeks 1–2), then once weekly
- Intrathecal methotrexate ± hydrocortisone: for patients with neurologic symptoms or abnormal CSF
HLH-2004 modified this by adding cyclosporine A (target trough 200 μg/L) from day 1 rather than week 9.
Continuation therapy / bridge to HSCT:
- Pulse dexamethasone every 2 weeks
- Alternating weekly etoposide
- Cyclosporine A throughout
Hematopoietic Stem Cell Transplantation (HSCT)
Allogeneic HSCT is the only curative therapy for primary HLH and is uniformly necessary in FHL (which is otherwise fatal). It should be performed as soon as an acceptable donor is identified, following remission induction. Reduced-intensity conditioning regimens have improved transplant-related mortality. In secondary HLH, HSCT is rarely needed but may be considered for refractory cases.
Targeted Biological Therapies
- Emapalumab (anti-IFN-γ monoclonal antibody): approved by the FDA for primary HLH in pediatric and adult patients refractory or intolerant to conventional therapy. Directly blocks the central pathogenic cytokine.
- Ruxolitinib (JAK1/2 inhibitor): increasingly used in both primary and secondary HLH; inhibits JAK-STAT signaling downstream of IFN-γ and IL-6. Particularly studied in MAS-HLH and adults with secondary HLH.
- Anakinra (IL-1Ra): used especially in MAS-HLH (SoJIA/AOSD-associated) where IL-1β and IL-18 are prominent drivers.
- Tocilizumab (anti-IL-6): used in some secondary HLH cases.
- IVIG: used as adjunctive therapy particularly in viral-triggered HLH.
Treatment by Trigger
| Trigger | Approach |
|---|---|
| EBV-HLH | Mild/moderate: steroids ± IVIG ± cyclosporin A; Severe: etoposide + dexamethasone; Add rituximab to clear B-cell EBV reservoir |
| Lymphoma-associated | Immediate chemotherapy (etoposide-containing regimen) + steroids |
| MAS (SoJIA/AOSD) | Anakinra, steroids, cyclosporin A; consider ruxolitinib for refractory cases |
| Sepsis/bacterial-associated | Treat infection first; add HLH-directed therapy if not responding |
| Primary FHL | HLH-94/2004 induction → HSCT |
| NLRC4 | IL-18 inhibition |
| XLP1 | Rituximab for EBV; HSCT early |
Prognosis
Without treatment, FHL is universally fatal with a median survival of 1–2 months. With the HLH-94 protocol and HSCT, 5-year survival in primary HLH approaches 54–65% in registries. CNS involvement at diagnosis is a major adverse prognostic factor; neurologic sequelae (psychomotor retardation ~25%, epilepsy ~10%, ADHD ~10%) persist in ~30% of survivors. Approximately 10% develop endocrinopathies.
Mortality in secondary HLH remains high — 20–50% depending on trigger — with malignancy-associated HLH carrying the worst prognosis. Early recognition is the single most modifiable determinant of outcome. — Harrison's 22E; Goldman-Cecil Medicine; Robbins & Kumar Basic Pathology
Summary
HLH is a paradigm of immunopathology in which a failure to terminate the immune response — rather than a failure to mount one — drives catastrophic tissue injury. The unifying pathophysiologic lesion across all forms is defective cytotoxic lymphocyte function leading to persistent antigen exposure, IFN-γ–mediated macrophage hyperactivation, and a self-amplifying cytokine storm. Recognition requires a high index of clinical suspicion, particularly in any patient with unexplained fever, organomegaly, cytopenias, and hyperferritinemia that does not respond to antibiotics. Prompt targeted therapy — increasingly guided by the underlying biology of each subtype — is essential to prevent death.
Sources: Harrison's Principles of Internal Medicine, 22E (2025) · Goldman-Cecil Medicine, International Edition · Robbins & Kumar Basic Pathology · Robbins, Cotran & Kumar Pathologic Basis of Disease
Give it as am answer short note question
Hemophagocytic Lymphohistiocytosis (HLH)
Definition
HLH is a life-threatening syndrome of uncontrolled immune activation characterized by massive proliferation of activated macrophages and CD8⁺ T lymphocytes causing a "cytokine storm," multiorgan failure, and death if untreated.
Classification
- Primary (Familial) HLH – autosomal recessive; caused by mutations in genes encoding cytolytic granule proteins (PRF1, UNC13D, STX11, STXBP2); mainly affects infants
- Secondary (Acquired) HLH – triggered by infection (EBV most common), malignancy (lymphoma, especially T/NK-cell), or autoimmune disease (SoJIA, SLE); termed Macrophage Activation Syndrome (MAS) in autoimmune context; mainly affects adults
Pathophysiology
Defective NK/CTL cytotoxicity → failure to clear antigen → persistent T-cell activation → IFN-γ–driven macrophage hyperactivation → hypercytokinemia (IFN-γ, IL-1β, IL-6, IL-18, TNF-α) → hemophagocytosis + organ damage
Clinical Features
| Feature | Detail |
|---|---|
| Fever | Prolonged, high-grade |
| Organomegaly | Hepatosplenomegaly (almost universal) |
| Cytopenias | Thrombocytopenia earliest; bicytopenia/pancytopenia |
| Liver | Elevated transaminases, jaundice, DIC, low fibrinogen |
| CNS | Seizures, altered consciousness (especially children) |
| Skin | Petechiae, purpura, maculopapular rash |
| Labs | ↑↑ Ferritin, ↑ triglycerides, ↓ fibrinogen, ↑ sCD25 |
Diagnosis — HLH-2004 Criteria
Molecular diagnosis of familial HLH gene OR ≥5 of 8:
- Fever
- Splenomegaly
- Bicytopenia (≥2 cell lines)
- Hypertriglyceridemia and/or hypofibrinogenemia
- Ferritin ≥500 μg/L
- Soluble CD25 (sIL-2r) ≥2400 U/mL
- Hemophagocytosis on bone marrow/spleen/lymph node biopsy
- Low/absent NK-cell activity
Ferritin >10,000 μg/L is highly suggestive. The HScore is an alternative tool validated for adults.
Treatment
| Setting | Treatment |
|---|---|
| Primary HLH | HLH-94/2004 protocol → HSCT (only cure) |
| HLH-94 induction | Dexamethasone + Etoposide (± intrathecal MTX for CNS disease) for 8 weeks |
| EBV-HLH | Dexamethasone + Etoposide + Rituximab |
| Malignancy-HLH | Immediate etoposide-based chemotherapy |
| MAS-HLH | Steroids + Cyclosporin A + Anakinra |
| Refractory HLH | Emapalumab (anti-IFN-γ); Ruxolitinib (JAK inhibitor) |
Prognosis
- Untreated FHL: median survival 1–2 months
- With HLH-94 + HSCT: 5-year survival ~54–65%
- Neurologic sequelae in ~30% of survivors
- Secondary HLH mortality: 20–50% depending on trigger
Sources: Harrison's Principles of Internal Medicine 22E · Goldman-Cecil Medicine · Robbins & Kumar Basic Pathology
Targeted therapies for AML
Reading File
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.