Ewing sarcoma
Reading File
Finding Sources
Searching PubMed
"Ewing sarcoma"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material to write a thorough answer. Let me compile everything.
Ewing Sarcoma
Overview
Ewing sarcoma is the third most common nonhematologic primary bone malignancy overall, but the second most common bone sarcoma in patients under 30 (after osteosarcoma) and the most common in patients under 10 years of age. It accounts for approximately 10-15% of all primary malignant bone tumors. Around 200 cases are diagnosed in the United States per year - incidence is less than 1 per million per year. There is a slight male predominance. Ewing sarcoma is exceedingly rare in individuals of African descent and occurs primarily in whites of European origin.
- Campbell's Operative Orthopaedics 15th Ed 2026, p. 1221
- Robbins & Kumar Basic Pathology, p. 786
- Goldman-Cecil Medicine, p. 1432
Molecular Pathogenesis
The defining feature of Ewing sarcoma is a balanced chromosomal translocation involving the EWSR1 gene on chromosome 22:
| Translocation | Genes fused | Frequency |
|---|---|---|
| t(11;22)(q24;q12) | EWSR1 - FLI1 | ~85% |
| t(21;22)(q22;q12) | EWSR1 - ERG | ~10% |
| t(7;22)(p22;q12) | EWSR1 - ETV1 | Rare |
-
90% of cases carry one of these EWS-ETS family fusions
- The chimeric EWS/FLI1 protein binds chromatin and dysregulates transcription, leading to uncontrolled growth and abnormal differentiation
- The cell of origin is uncertain - mesenchymal stem cells and primitive neuroectodermal cells are the leading candidates
The specific translocation type (t(11;22) vs. t(21;22)) does not seem to affect the clinical course, though secondary genetic alterations such as aberrant TP53 expression may carry prognostic significance.
- Robbins & Kumar Basic Pathology, p. 786
- Miller's Review of Orthopaedics 9th Ed
- Campbell's Operative Orthopaedics 15th Ed 2026, p. 1222
Epidemiology & Location
- Age: Most cases ages 5-25 years; peak in the second decade. About 20% occur in older patients.
- Sex: Slight male predominance
- Sites: Metaphyses of long bones (with frequent extension into the diaphysis), flat bones of the shoulder and pelvic girdles. Classic teaching is "diaphyseal" but metaphyseal origin is actually more common, with diaphyseal extension.
- Also: pelvis, ribs, sacrum (vertebral Ewing sarcoma accounts for 3.3-15% of cases, with sacral involvement in up to 50%)
- Extraskeletal: ~20% of Ewing sarcomas are extraskeletal ("extraosseous Ewing sarcoma")
- Rare: spine, small bones of the hands/feet
Clinical Presentation
Pain is almost universal. Key features:
- Insidious onset with long duration before diagnosis (average delay = 34 weeks)
- Patient delay: ~15 weeks to first appointment
- Physician delay: ~19 weeks from first visit to diagnosis
- Pain may initially be mild/intermittent and respond to conservative treatment
- Fever, erythema, and swelling - can mimic osteomyelitis (a notorious diagnostic pitfall)
- Lab abnormalities: elevated WBC, elevated ESR, elevated CRP, elevated LDH (a prognostic marker)
- A needle aspirate may grossly resemble pus - always send specimen for both culture and pathology
Imaging
Plain Radiograph
- Classically: destructive diaphyseal lesion with "onion skin" periosteal reaction (layers of reactive bone)
- Also: permeative / "moth-eaten" appearance
- A large portion of the bone or even the entire bone may be involved
- Flat bones show a nonspecific destructive appearance
- Associated soft-tissue mass is often very large
MRI (modality of choice for staging)
- Order for the entire bone - extent typically exceeds what is apparent on plain films
- Evaluates the soft-tissue component
- Vertebral Ewing: usually isointense to hyperintense on T2; isointense on T1; intense gadolinium enhancement ("curtain sign" on axial images for spinal canal invasion)
Staging workup
| Study | Purpose |
|---|---|
| Chest X-ray + CT | Lung - most common metastatic site |
| Bone scan | Bone - second most common metastatic site |
| FDG-PET/CT | High sensitivity for staging, detecting recurrence; new standard; initial SUV correlates with prognosis |
| Bone marrow aspirate | Rule out diffuse systemic disease |
| Whole-body MRI | Alternative to bone scan/PET |
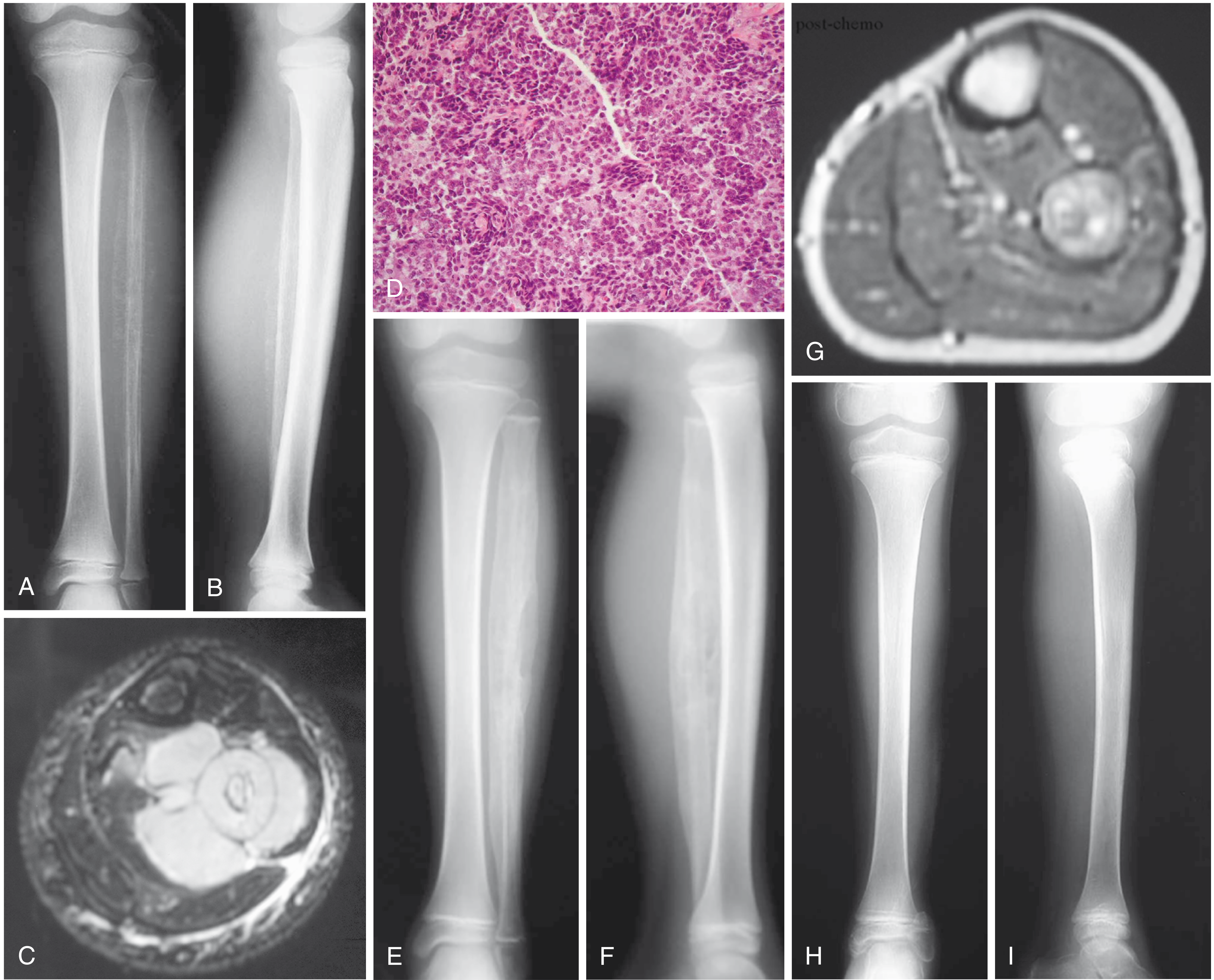
Fig: (A,B) AP and lateral radiographs of left fibula of a 7-year-old girl with Ewing sarcoma. (C) MRI shows large soft-tissue mass. (D) Typical microscopic appearance. (E,F) After neoadjuvant chemotherapy - increased ossification. (G) Post-chemo MRI showing marked reduction in soft-tissue mass. (H,I) After wide resection. - Campbell's Operative Orthopaedics 15th Ed 2026
Histopathology
Ewing sarcoma belongs to the small round blue cell tumors of childhood. Key histological features:
- Sheets of uniform, small round cells slightly larger than lymphocytes
- Minimal intercellular matrix - no bone or cartilage production (distinguishes from osteosarcoma)
- Scant cytoplasm, may appear clear due to glycogen content
- Usually arises in the medullary cavity, invades cortex, periosteum, and soft tissue
- Homer-Wright rosettes (circular cell groupings with a central fibrillary core) may be present
Special stains and IHC
| Marker | Ewing sarcoma | Lymphoma |
|---|---|---|
| PAS stain | Positive (glycogen) | Negative |
| Reticulin | Negative | Positive |
| MIC-2 (CD99) | Positive - specific for Ewing | Negative |
| Leukocyte common antigen (CD45) | Negative | Positive |
Cytogenetic or IHC studies are usually required to distinguish Ewing sarcoma from other small blue cell tumors (lymphoma, rhabdomyosarcoma, neuroblastoma, small cell carcinoma).
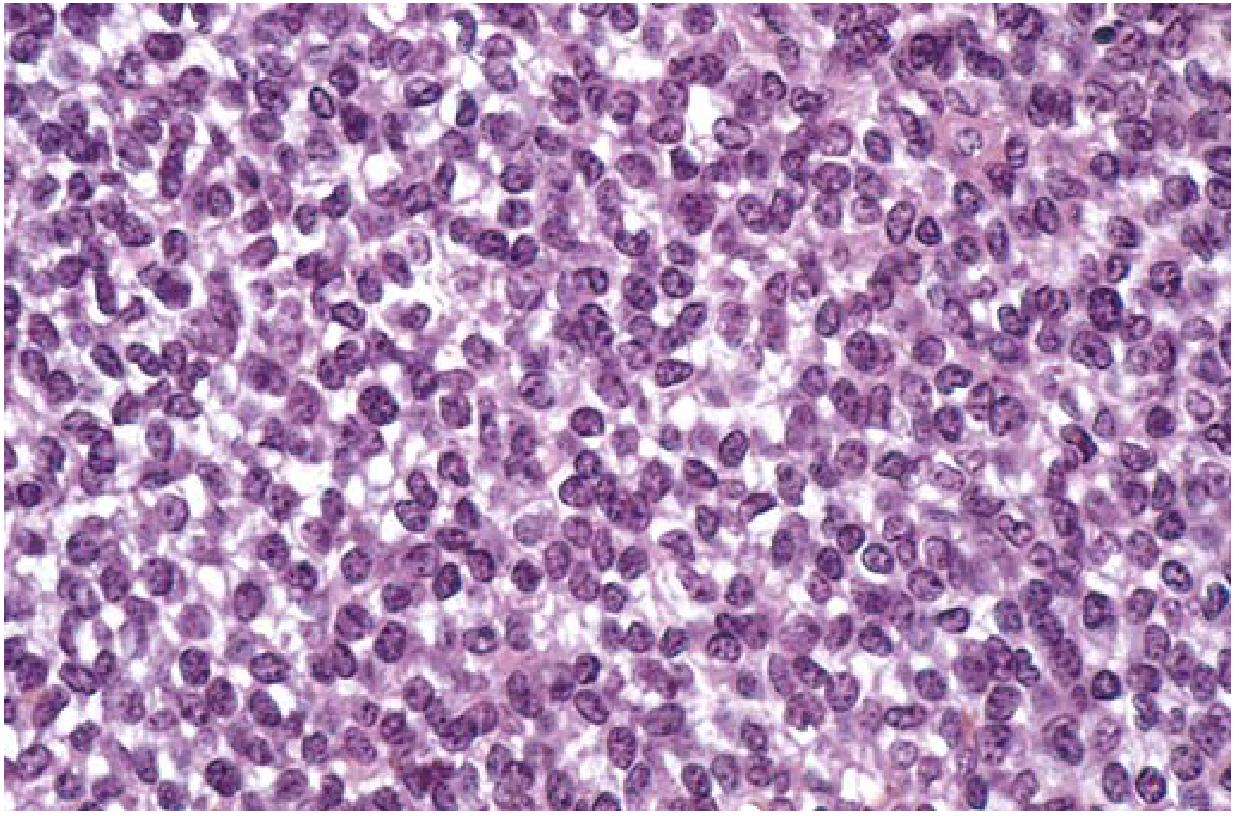
Fig: Ewing sarcoma composed of sheets of small round cells with minimal clear cytoplasm. H&E stain. - Robbins & Kumar Basic Pathology
Treatment
Treatment is multimodal and must always include systemic chemotherapy because micrometastatic disease is presumed present at diagnosis.
Chemotherapy
- Neoadjuvant + adjuvant chemotherapy is the cornerstone
- Standard regimen: VDC/IE - vincristine, doxorubicin, cyclophosphamide (VDC) alternating with ifosfamide and etoposide (IE), administered under expert guidance with dose intensification
- Salvage regimens for relapse: cyclophosphamide/topotecan, irinotecan/temozolomide, gemcitabine/docetaxel
Local Control (Surgery vs. Radiation)
The choice is individualized:
| Option | Considerations |
|---|---|
| Wide surgical resection | Preferred when achievable with acceptable functional deficit; local recurrence rate <10%; avoids radiation-related growth disturbances in children |
| Radiation therapy | Ewing sarcoma is radiosensitive; used for large, central, unresectable tumors (e.g., pelvis); adjuvant after marginal or contaminated resection |
| Combination | Radiation as adjuvant after surgery if margins are inadequate |
Repeat staging (radiographs + MRI) is obtained after neoadjuvant chemotherapy before deciding on local treatment. Response is often dramatic - increased ossification on X-ray and marked reduction of soft-tissue mass on MRI.
Prognosis
Overall Survival
- With modern multiagent chemotherapy: 5-year event-free survival ~70% (localized disease)
- Before multiagent chemotherapy: <10% long-term survival
- Current centers report 60-75% long-term survival for localized disease
Poor Prognostic Factors
| Factor | Comment |
|---|---|
| Distant metastases at presentation | Most important; 20-30% long-term survival despite aggressive treatment |
| Large primary tumor size | Consistently significant |
| Proximal/axial location | Often reflects larger tumor size |
| Elevated LDH, WBC, ESR | Indicates more extensive disease |
| Fever and anemia | More extensive disease |
| Older age (>12-15 years) | Worse prognosis |
| Male gender | Slight disadvantage |
| Aberrant TP53 expression | Secondary genetic alteration with adverse impact |
| Poor histologic response to neoadjuvant chemo | <90% necrosis = poor response |
Good Prognostic Factor
- >90% necrosis after neoadjuvant chemotherapy = good histologic response and favorable prognosis
Recurrence
- Local recurrence: ~20% 5-year survival
- Distant relapse: ~10% 5-year survival
- Relapse within the first year carries a worse prognosis than late relapse
Differential Diagnosis (Small Round Blue Cell Tumors in Children)
- Lymphoma - PAS negative, reticulin positive, LCA positive
- Rhabdomyosarcoma - desmin, myoglobin, muscle-specific actin positive
- Neuroblastoma - paraspinal, adrenal; urinary catecholamines elevated
- Small cell carcinoma - cytokeratin positive
- Osteomyelitis - can be clinically indistinguishable (both cause pain, fever, elevated inflammatory markers, pus-like aspirate)
Key sources: Campbell's Operative Orthopaedics 15th Ed 2026, pp. 1221-1223; Robbins & Kumar Basic Pathology, pp. 786-787; Goldman-Cecil Medicine, pp. 1432-1443; Grainger & Allison's Diagnostic Radiology, p. 1285
This is a shared conversation. Sign in to Orris to start your own chat.