Osteoarthritis
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"osteoarthritis"[MeSH Terms] AND "pathophysiology"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive overview of osteoarthritis covering definition, pathophysiology, and clinical features.
Osteoarthritis (OA)
Definition
Osteoarthritis — also called degenerative joint disease — is characterized by failed repair of synovial joint tissues after intra-articular stress, resulting in breakdown of cartilage and bone that leads to pain, stiffness, and functional disability. Although the name implies inflammation, it is primarily a degenerative disorder of articular cartilage, with inflammation acting as a secondary contributor.
Goldman-Cecil Medicine, p. 2766; Robbins & Kumar Basic Pathology, p. 790
Epidemiology
| Fact | Detail |
|---|---|
| Global prevalence | >500 million people worldwide |
| Sex | Women disproportionately affected |
| Age | Incidence rises sharply between ages 50–75; ~40% of people >70 affected |
| Projected burden | ~25% of U.S. adults expected to have physician-diagnosed OA by 2040 |
| Disability | 80% of those affected have movement limitation; 25% have difficulty with ADLs |
| Knee OA | Accounts for ~85% of global OA burden; lifetime risk ~40% (men) and ~47% (women) |
The number of people affected globally rose by 48% from 1990 to 2019, driven largely by aging, obesity, and joint injury. OA is now the 15th highest cause of years lived with disability worldwide.
Classification
| Type | Description |
|---|---|
| Primary (idiopathic) | Appears insidiously with aging; oligoarticular, affecting weight-bearing joints |
| Secondary | ~5% of cases; younger patients with predisposing condition — prior joint injury, deformity, diabetes, obesity |
Pathophysiology
OA results from a dynamic imbalance between repair and destruction of joint tissues, driven by mechanical, inflammatory, and metabolic pathways.
Stage 1 — Chondrocyte Injury
Biomechanical stress (the principal mechanism) or genetic predisposition triggers chondrocyte injury. Polymorphisms in matrix components and signaling molecules predispose individuals; over 100 DNA variants with polygenic effects have been identified, accounting for >20% of OA heritability.
Stage 2 — Early OA (Repair attempt fails)
- Injured chondrocytes proliferate in an attempt to repair matrix loss
- They secrete matrix metalloproteinases (MMPs) that degrade type II collagen and proteoglycans
- Proinflammatory mediators released: PGE₂, nitric oxide (NO), TNF
- TGF-β and BMPs are also generated — attempting repair, but degradation exceeds it
- Cartilage initially swells as proteoglycans attract water, then the type II collagen matrix disrupts
- Breakdown products stimulate the synovium to become hyperplastic (more lining cells) and hypertrophic (villi with macrophage and lymphocyte infiltration)
Stage 3 — Late OA
- Full-thickness loss of cartilage, chondrocyte apoptosis and dropout
- Dislodged fragments become loose bodies ("joint mice")
- Exposed subchondral bone becomes the new articular surface and is burnished to a polished ivory appearance → bone eburnation
- Fractures in subchondral bone allow synovial fluid forced in by a ball-valve mechanism → subchondral cysts
- Reactivation of endochondral ossification at joint margins forms osteophytes (bony spurs)
- Increased bone turnover with subchondral bone marrow lesions — associated with both pain and disease progression
- Meniscal degeneration, ligamentous laxity, and periarticular muscle atrophy are common
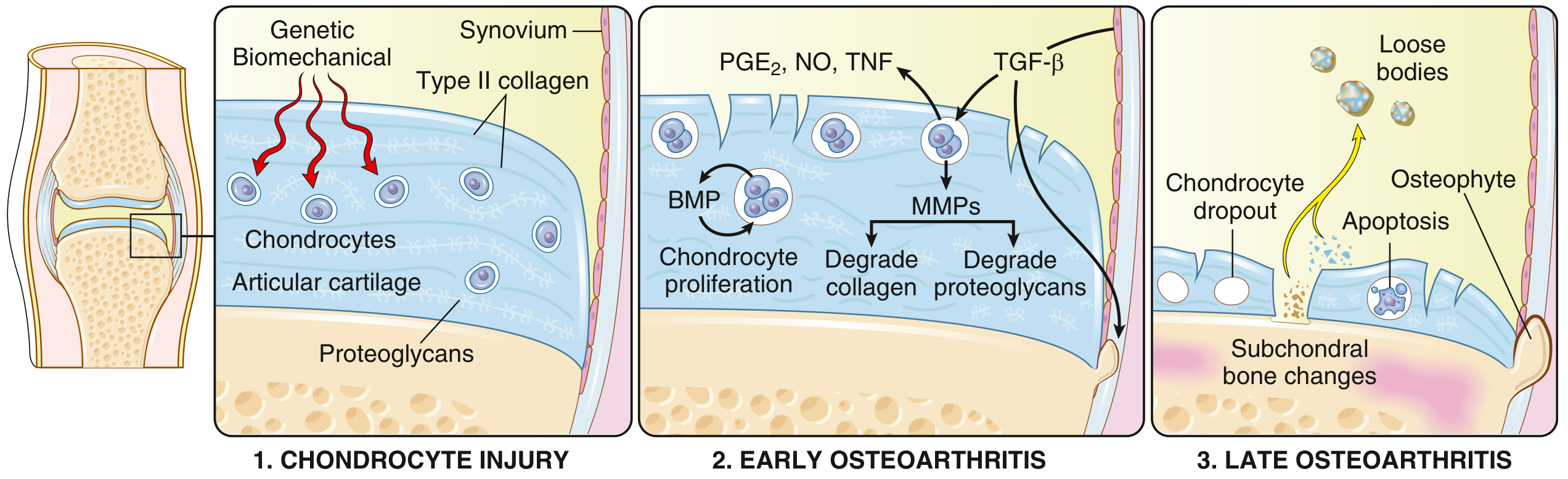
Mechanics vs. Inflammation
Mechanical load is the primary risk factor. Excessive strain through a normal joint (obesity, malalignment, occupational overload) OR normal load through a structurally compromised joint (torn meniscus, weak muscles, ligamentous laxity) activates inflammatory signaling pathways. Systemic, obesity-driven inflammation also perpetuates the process.
Risk Factors
| Modifiable | Non-modifiable |
|---|---|
| Increased body weight | Age |
| Joint malalignment | Sex (female) |
| Muscle weakness | Ethnicity |
| Sedentary lifestyle | Genetics |
| Occupational exposure | Prior injury |
| Athletic injury |
Morphology / Gross & Histological Changes
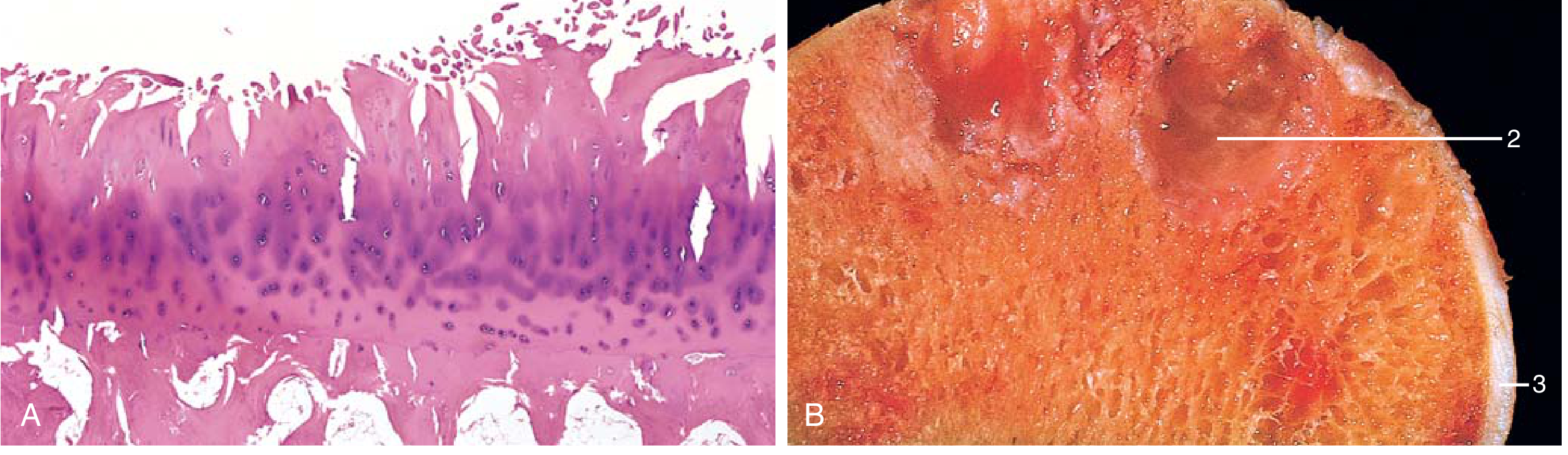
| Finding | Description |
|---|---|
| Fibrillation | Superficial splitting/fraying of cartilage (earliest histologic change) |
| Full-thickness cartilage loss | Sloughing exposes subchondral bone |
| Eburnation | Polished ivory appearance of exposed bone |
| Subchondral cysts | Fluid-filled, fibrous-walled cavities in bone |
| Osteophytes | Bony outgrowths at articular margins, capped by fibrocartilage |
| Loose bodies | Detached cartilage/bone fragments within joint space |
| Synovium | Mild congestion and fibrosis; scattered chronic inflammatory cells |
Etiology of Pain
Pain in OA is understood through the biopsychosocial model. Three mechanisms operate:
- Peripheral nociceptive pain — mechanical loading of a damaged joint activates joint nociceptors
- Neuropathic/central sensitization — altered neurophysiologic signaling amplifies pain; these patients often respond poorly to standard treatments
- Subchondral bone marrow lesions — strongly associated with pain severity
Importantly, the degree of structural OA correlates only moderately with pain severity — imaging findings can be severe with minimal symptoms, and vice versa.
Clinical Features
Symptoms
| Feature | Detail |
|---|---|
| Pain | Gradual onset; mechanical in nature; worsens with activity; worse towards end of day; at rest in advanced disease |
| Morning stiffness | Localized to involved joint; usually <30 minutes (cf. RA >1 hour) |
| Crepitus | Grating/crackling sensation on joint movement |
| Limited range of motion | Progressive |
| Joint deformity | Occurs with time; joint fusion does not occur (unlike RA) |
Signs / Characteristic Physical Findings
| Finding | Joint |
|---|---|
| Heberden nodes | Osteophytes at distal interphalangeal (DIP) joints — more common in women |
| Bouchard nodes | Osteophytes at proximal interphalangeal (PIP) joints |
| Radicular pain, muscle spasm, neurologic deficits | Osteophyte impingement on spinal foramina |
| Bony enlargement, reduced ROM | Knees, hips |
Joints Commonly Affected
- Hips, knees (weight-bearing)
- Lower lumbar and cervical vertebrae
- DIP and PIP joints of fingers
- First carpometacarpal joints
- First tarsometatarsal joints
OA Phenotypes (Knee)
- Chronic pain phenotype
- Inflammatory phenotype
- Metabolic syndrome phenotype
- Bone and cartilage metabolism phenotype
- Mechanical overload phenotype
- Minimal joint disease phenotype
OA vs. Rheumatoid Arthritis — Key Distinctions
| Feature | OA | RA |
|---|---|---|
| Primary mechanism | Mechanical/degenerative | Autoimmune |
| Inflammation | Secondary | Primary |
| Joints | Weight-bearing (hips, knees) | Small joints of fingers first |
| Stiffness | <30 min | >1 hour (morning) |
| Serum antibodies | None | ACPA, rheumatoid factor |
| Joint fusion | No | Yes (ankylosis) |
| Extra-articular involvement | No | Yes (lungs, heart, etc.) |
Sources: Robbins & Kumar Basic Pathology, pp. 790–791 | Goldman-Cecil Medicine, pp. 2765–2770
Recent literature note: A 2024 systematic review on OA biomechanics (PMID 39369839) and a 2026 review on macrophage polarization in OA (PMID 41412012) are consistent with the pathophysiologic framework above, with emerging research highlighting the role of immune cells (T-cell receptor repertoire, macrophage polarization) in disease progression — PMID 40912449.
This is a shared conversation. Sign in to Orris to start your own chat.