Polycythemia vera
Reading File
Finding Sources
Searching PubMed
"polycythemia vera"[MeSH Terms]
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
I have rich, comprehensive content from multiple authoritative textbooks. Now let me compile the full answer.
Polycythemia Vera (PV)
Definition & Classification
Polycythemia vera (PV) is a clonal myeloproliferative neoplasm (MPN) arising from a transformed hematopoietic stem cell. It is classified as a Philadelphia chromosome-negative (Ph-negative) MPN alongside essential thrombocythemia (ET) and primary myelofibrosis (PMF). The hallmark is panmyelosis - excessive proliferation of erythroid, granulocytic, and megakaryocytic elements - but it is the absolute increase in red cell mass that drives most clinical manifestations.
-
Incidence: 1-3 per 100,000/year in the US, increasing with age to >10/100,000 in older adults
-
Familial transmission is uncommon; sporadic cases show female predominance under age 50
-
Harrison's Principles of Internal Medicine, 22E, p. 862
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 14
Pathogenesis
The JAK2 Mutation
The central molecular event is a point mutation in the JAK2 gene (chromosome 9p):
- JAK2 V617F (valine → phenylalanine at codon 617): found in >95-97% of PV patients. It sits in the autoinhibitory pseudokinase domain, causing constitutive, ligand-independent kinase activation.
- JAK2 normally serves as the cognate kinase for the erythropoietin receptor (EPOR) and thrombopoietin receptor. Constitutive activation bypasses the need for erythropoietin, explaining:
- Erythropoietin hypersensitivity and EPO-independent erythroid colony formation
- Increased Bcl-X(L) expression and apoptosis resistance in erythroid progenitors
- Reduced serum erythropoietin (a key diagnostic clue - opposite of secondary polycythemia)
- Exon 12 JAK2 mutations account for most remaining JAK2 V617F-negative cases - these tend to present with isolated erythrocytosis
- Loss of heterozygosity at 9p (uniparental disomy → JAK2 V617F homozygosity) occurs in ~60% of PV patients and is associated with higher WBC counts, more significant splenomegaly, pruritus, and faster progression to spent phase
- Rare additional driver mutations: LNK (a JAK2 inhibitor), CALR, occasional ASXL1, TET2, DNMT3A (clonal evolution markers)
The JAK2 V617F mutation is not the sole initiating lesion - the predisposition is associated with a specific JAK2 haplotype (GGCC haplotype), and familial PV can occur without the mutation.
- Harrison's, p. 862-863
- Robbins & Kumar Basic Pathology, p. 406-407
Morphology
Bone marrow: Hypercellular with panmyelosis (increased erythroid, myeloid, and megakaryocytic forms). Some residual fat is usually preserved. Moderate-to-marked reticulin fibrosis at diagnosis in ~10% of cases.
Late / "spent phase": Marrow replaced by fibroblasts and collagen (post-PV myelofibrosis). Extramedullary hematopoiesis in spleen and liver → prominent organomegaly.
Spleen: Mildly enlarged early (vascular congestion). Later, massive splenomegaly from extramedullary hematopoiesis.
Blood vessels: Thromboses and infarctions common in heart, spleen, kidneys; hemorrhages in ~30%.
Peripheral blood: RBC 6-12 × 10¹²/L; platelet count often >400,000/µL; WBC up to 50,000/µL; basophilia characteristic; giant platelets and megakaryocyte fragments; low/absent ESR (high viscosity).
- Robbins, Cotran & Kumar, p. 14-15
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 14
Clinical Features
Symptoms from Erythrocytosis / Hyperviscosity
- Neurological: Headache, vertigo, tinnitus, visual disturbances, transient ischemic attacks (TIAs), paresthesias
- Systemic hypertension
- Facial plethora, ruddy cyanosis, conjunctival injection, retinal vein engorgement
Thrombosis (most significant complication - ~30% of patients)
- Cerebral, cardiac, mesenteric vessels most common
- Hepatic vein thrombosis (Budd-Chiari syndrome) - especially in young women; PV should be suspected in any woman who develops this
- Portal vein thrombosis more common in males
- Arterial thrombosis in 16%, venous thrombosis in 7% at or before diagnosis
Hemorrhage (5-10% life-threatening)
- Epistaxis, gum bleeding, GI hemorrhage (often peptic ulcer)
- Paradoxically caused by platelet dysfunction and acquired von Willebrand disease (when platelet count >900,000/µL, large platelet mass absorbs high-MW vWF multimers)
Pruritus
- Aquagenic pruritus (itching after warm bath/shower) - affects 30-50% of patients; may precede PV diagnosis by years
- Due to histamine release from basophils
Erythromelalgia
- Erythema, burning, and pain in distal extremities (hands/feet) from platelet-mediated microvascular occlusion
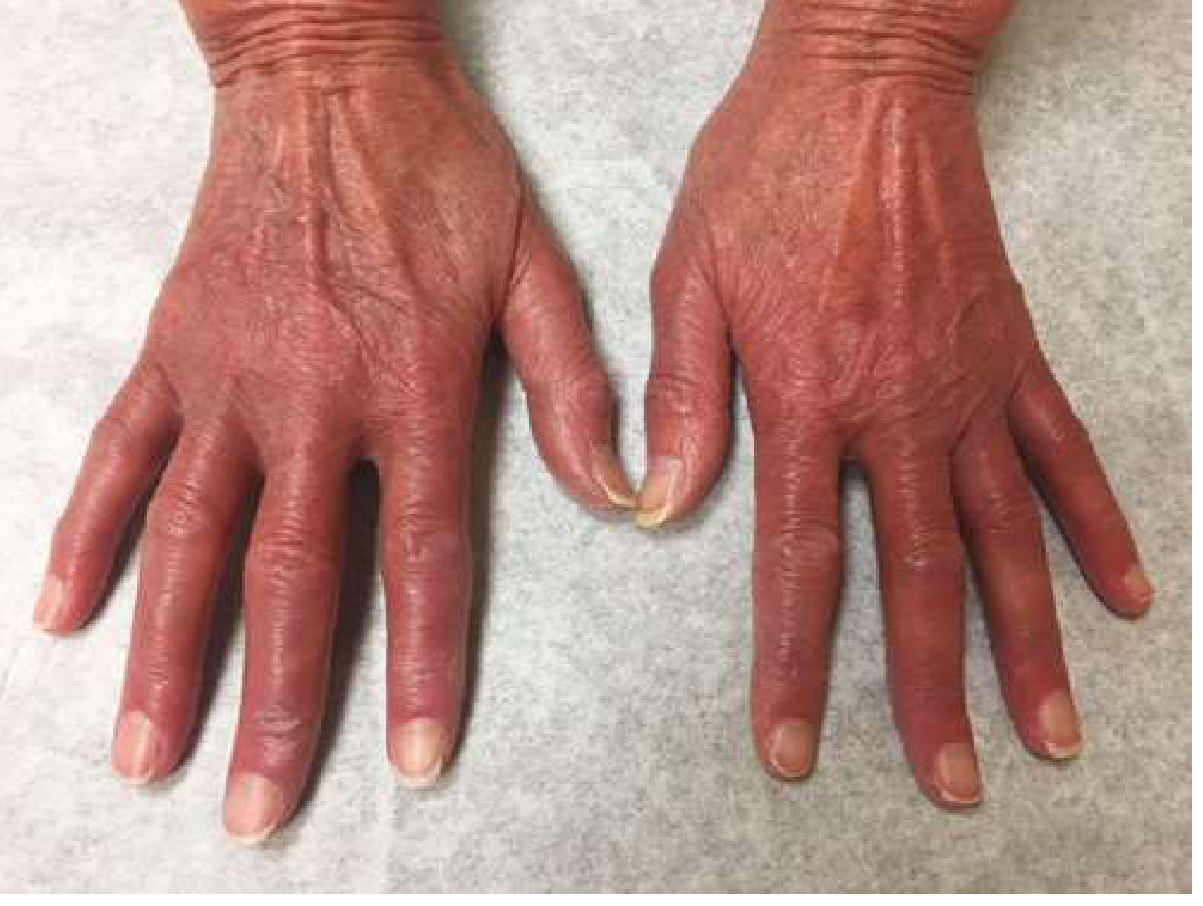
Gout / Hyperuricemia
- Symptomatic gout in 5-10% from high cell turnover and nucleic acid catabolism; uric acid stones
Splenomegaly
-
Present in two-thirds of patients at diagnosis
-
Harrison's, p. 862-863; Goldman-Cecil Medicine, p. 1750-1751; Henry's, p. 14
Laboratory Findings
| Parameter | Finding |
|---|---|
| Hemoglobin | >18.5 g/dL (men) / >16.5 g/dL (women) |
| Hematocrit | Often ≥60%; diagnostic threshold >49% men, >48% women |
| RBC count | 6-10 million/µL (up to 12 × 10¹²/L) |
| Serum EPO | Low (key diagnostic distinction from secondary polycythemia) |
| WBC | 10,000-50,000/µL; neutrophilia + basophilia |
| Platelets | >400,000/µL; often >1,000,000/µL; functionally abnormal |
| Neutrophil alkaline phosphatase (NAP) | Elevated in 80% |
| Uric acid | Elevated |
| ESR | Reduced (high viscosity) |
| Arterial O₂ saturation | Normal |
| Serum B12 | Often elevated (transcobalamin release from granulocytes) |
| JAK2 V617F mutation | Positive in >95% |
WHO Diagnostic Criteria (2016/2022)
Major criteria:
- Hemoglobin >16.5 g/dL (men) or >16.0 g/dL (women), OR hematocrit >49% (men) or >48% (women), OR increased red cell mass
- Bone marrow biopsy showing hypercellularity with panmyelosis (trilineage growth: erythroid, granulocytic, megakaryocytic proliferation) with pleomorphic mature megakaryocytes
- Presence of JAK2 V617F or JAK2 exon 12 mutation
Minor criterion:
- Subnormal serum EPO level
Diagnosis requires: All 3 major criteria, OR major criteria 1 + 2 with the minor criterion.
- Goldman-Cecil Medicine, p. 1750; Henry's, p. 14
Differential Diagnosis
PV must be distinguished from:
- Relative (spurious) polycythemia (Gaisböck's syndrome): plasma volume contraction from dehydration, diuretics, tobacco, androgens - no absolute increase in red cell mass
- Secondary absolute polycythemia: EPO levels are HIGH (opposite of PV):
- Hypoxia: COPD, high altitude, sleep apnea, high-affinity hemoglobin, carbon monoxide
- Ectopic EPO: hypernephroma, hepatocellular carcinoma, cerebellar hemangioblastoma, uterine fibroids
- Other MPNs: ET, PMF (can have same JAK2 V617F but different phenotype)
- CML (BCR-ABL1 positive - must be excluded)
Key distinguishing features of PV vs secondary polycythemia:
| Feature | PV | Secondary |
|---|---|---|
| Serum EPO | Low | High |
| Splenomegaly | Common | Absent |
| Leukocytosis/thrombocytosis | Yes | No |
| JAK2 mutation | >95% | No |
| Panmyelosis on BM | Yes | No |
Disease Course & Complications
Three phases:
- Proliferative phase: Controlled erythrocytosis, manageable with phlebotomy
- Stable phase: Iron deficiency limits red cell expansion; phlebotomy every 3 months
- Spent (post-PV MF) phase: Marrow fibrosis, cytopenias, massive splenomegaly, extramedullary hematopoiesis - occurs in a subset
Transformation:
- Post-PV myelofibrosis: variable frequency, higher with JAK2 V617F homozygosity
- AML transformation: ~1-2% without myelosuppressive therapy; higher (up to 10-15%) with alkylating agents or ³²P. Alkylating agents and radioactive phosphorus are leukemogenic and should be avoided
Survival: Without treatment, death from vascular complications within months. Median survival ~10 years with treatment (phlebotomy to maintain normal red cell mass).
- Robbins, Cotran & Kumar, p. 15; Harrison's, p. 864
Management
1. Phlebotomy (first-line for all patients)
- Goal: Hematocrit <45% in men, <42% in women (hematocrit ≥45% significantly increases thrombosis risk)
- Induces iron deficiency that limits re-expansion of red cell mass
- Once iron-deficient, phlebotomy usually needed only every 3 months
2. Low-dose Aspirin
- Reduces risk of thrombotic complications
- Caution: potentially harmful if red cell mass not controlled, and increases bleeding in patients >60 years
3. Cytoreductive Therapy (for high-risk patients)
Risk stratification:
- Low risk: Age <60, no thrombosis history
- High risk: Age ≥60 OR prior thrombosis
High-risk patients require cytoreduction in addition to phlebotomy:
| Drug | Notes |
|---|---|
| Hydroxyurea | Most widely used; effective but mutagenic (does not target the malignant HSC directly); leukemogenic with long use |
| Ruxolitinib (JAK1/2 inhibitor) | Targets involved HSCs; not mutagenic; reduces splenomegaly and pruritus; superior to best available therapy in trials |
| Pegylated IFN-α (ropeginterferon) | Targets malignant HSCs; achieves complete hematologic AND molecular remissions in ~20%; preferred in younger patients; biweekly dosing |
| Busulfan | Used in elderly; avoid alkylating agents for long-term use (leukemogenic) |
| ³²P (radioactive phosphorus) | Effective but leukemogenic; rarely used now |
4. Symptom Management
- Pruritus: Antihistamines, antidepressants (doxepin), ruxolitinib, pegIFN-α, hydroxyurea, PUVA
- Hyperuricemia/gout: Allopurinol (especially when starting chemotherapy)
- Erythromelalgia: Low-dose aspirin (usually very effective); platelet count reduction if needed
- Thrombosis: Anticoagulation; newer oral anticoagulants preferred over VKA (no monitoring required)
- Splenomegaly: Ruxolitinib or pegIFN-α; splenectomy rarely needed now with effective JAK2 inhibitors
5. Newer/Emerging Agents
- Ropeginterferon alfa-2b (Besremi): long-acting pegylated interferon; approved specifically for PV. A 2026 systematic review and meta-analysis confirms significant hematologic and molecular response rates.
- A 2024 meta-analysis confirmed ruxolitinib superior to best available therapy for hematocrit control, symptom burden, and spleen volume reduction in hydroxyurea-intolerant/resistant PV.
Key Points Summary
| Feature | Detail |
|---|---|
| Mutation | JAK2 V617F (>95%); exon 12 for most remainder |
| Mechanism | Constitutive JAK/STAT activation → EPO-independent erythropoiesis |
| EPO level | Low (key diagnostic finding) |
| Classic symptom | Aquagenic pruritus, erythromelalgia, plethora |
| Biggest complication | Thrombosis (cerebral, cardiac, Budd-Chiari) |
| Diagnosis | WHO criteria: panmyelosis + elevated Hgb/Hct + JAK2 mutation ± low EPO |
| First-line Rx | Phlebotomy to Hct <45% + low-dose aspirin |
| High-risk Rx | Add ruxolitinib or pegIFN-α (preferred) or hydroxyurea |
| Avoid | Alkylating agents, ³²P (leukemogenic) |
| Prognosis | Median survival ~10 years with treatment |
Sources: Harrison's Principles of Internal Medicine, 22E (2025); Robbins, Cotran & Kumar Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Goldman-Cecil Medicine; Henry's Clinical Diagnosis and Management by Laboratory Methods
This is a shared conversation. Sign in to Orris to start your own chat.