Heart failure
Reading File
Asking for Preferences
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive content to deliver a well-organized, exam-focused overview. Here is the full answer:
Heart Failure - Overview for Medical Students
Definition
Heart failure (HF) is a clinical syndrome in which the heart cannot pump blood to adequately meet the metabolic demands of peripheral tissues, or can do so only at elevated filling pressures. It is the common final pathway of many chronic heart diseases.
- Robbins Pathologic Basis of Disease - affects >6.5 million in the US (~2% of population), ~50% mortality within 5 years of diagnosis
- Textbook of Family Medicine 9e - preferred over the older term "congestive heart failure" because pulmonary congestion is not always present
Types of Heart Failure
By Ejection Fraction (most important classification)
| Type | EF | Mechanism |
|---|---|---|
| HFrEF (Heart Failure with Reduced EF) | ≤40% | Systolic dysfunction - impaired LV contraction |
| HFmrEF (mildly reduced EF) | 41-49% | Mixed features |
| HFpEF (Heart Failure with Preserved EF) | ≥50% | Diastolic dysfunction - impaired LV filling/relaxation |
HFpEF patients tend to be older, female, obese, and diabetic. HFpEF and HFrEF are roughly equally common.
By Side
| Type | Cause | Consequences |
|---|---|---|
| Left-sided HF | IHD, hypertension, aortic/mitral valve disease, cardiomyopathy | Pulmonary congestion - dyspnoea, orthopnoea, PND, pulmonary oedema |
| Right-sided HF | Most commonly from left-sided HF (via pulmonary hypertension); also cor pulmonale | Systemic venous congestion - JVD, hepatomegaly, peripheral oedema, ascites |
By Onset
- Acute HF - sudden decompensation (e.g. acute MI, flash pulmonary oedema)
- Chronic HF - gradual progression with compensatory mechanisms
By Output
- Low-output HF - most common; heart cannot generate adequate CO
- High-output HF - paradoxically elevated CO but still cannot meet excessive demand (causes: thyrotoxicosis, Paget disease, anaemia, AV fistulae, beriberi)
Compensatory Mechanisms (Initially Helpful, Ultimately Harmful)
When cardiac function is compromised, three major adaptive responses activate:
-
Frank-Starling mechanism - increased filling volumes stretch myocytes, increasing actin-myosin cross-bridge formation for a more forceful contraction. This is short-term effective but chronic volume overload causes dilation and failure.
-
Neurohumoral activation:
- SNS: Norepinephrine release - increases HR, contractility, and vascular resistance
- RAAS: Renin-Ang II-aldosterone axis - promotes salt/water retention, vasoconstriction
- ANP/BNP: Released by stretched atria/ventricles as a counter-regulatory response - promotes diuresis and vasodilation
-
Cardiac hypertrophy / LV remodeling - the heart increases myocyte mass to handle increased wall stress. Over time this leads to adverse remodeling: dilation, fibrosis, and progressive dysfunction.
Pathophysiology: The Neurohormonal Model
The old "hemodynamic model" has been replaced by the LV remodeling/neurohormonal model.
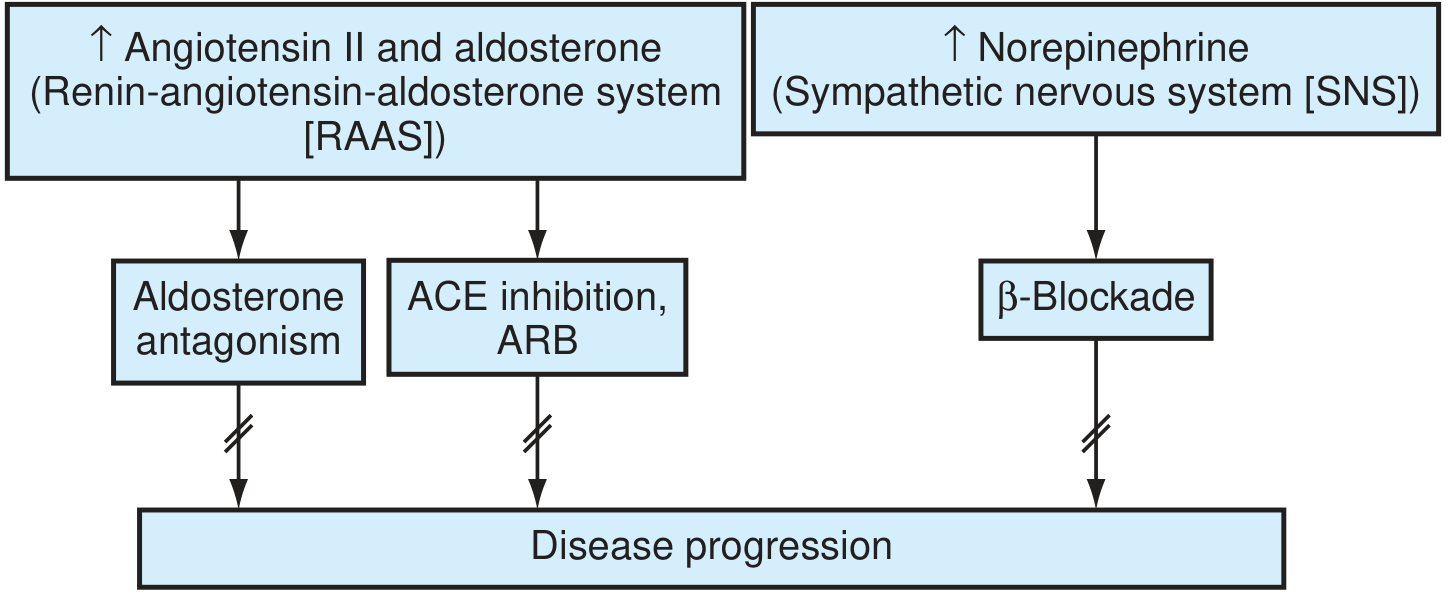
The RAAS and SNS drive disease progression; blocking them is the cornerstone of treatment. - Textbook of Family Medicine 9e
Key pathological cascades:
- Angiotensin II promotes cardiomyocyte apoptosis, hypertrophy, and ventricular fibrosis; stimulates aldosterone secretion
- Aldosterone "escapes" ACE inhibitor suppression over time, independently promoting adverse remodeling (explains the need for MRA therapy)
- Catecholamines downregulate adrenergic receptors, cause Ca²⁺ overload in myocytes, increase O₂ consumption, and trigger arrhythmias
- Endothelin-1 (from dysfunctional endothelium) causes vasoconstriction
- Inflammatory cytokines (TNF-α, IL-6) further worsen endothelial function
- MMPs (matrix metalloproteinases) promote cardiac fibrosis and collagen deposition
- Metabolic shift - from fatty acid oxidation to glycolysis (less efficient)
Goodman & Gilman summarizes four major interrelated systems: the heart itself, the vasculature, the kidneys, and neurohumoral circuits.
Common Causes (by type)
HFrEF:
- Ischaemic heart disease / MI (most common)
- Dilated cardiomyopathy
- Hypertension (long-standing)
- Valvular disease (aortic regurgitation, mitral regurgitation)
- Myocarditis
HFpEF:
- Hypertension (LV hypertrophy)
- Obesity / metabolic syndrome
- Diabetes mellitus
- Constrictive pericarditis
- Amyloidosis
- Hypertrophic cardiomyopathy
Forward vs. Backward Failure
| Concept | Mechanism | Result |
|---|---|---|
| Forward failure | Decreased cardiac output / tissue perfusion | Fatigue, weakness, renal hypoperfusion (pre-renal azotaemia), cardiogenic shock |
| Backward failure | Blood pools in venous capacitance system | Left-sided: pulmonary oedema; Right-sided: peripheral oedema, hepatomegaly, ascites |
Key Exam Points
- HF ≠ cardiomegaly, and it ≠ always = pulmonary congestion
- BNP/NT-proBNP is elevated in both HFrEF and HFpEF - useful biomarker for diagnosis and prognosis
- The ejection fraction is the single most important prognostic and management-guiding parameter
- LV remodeling is reversible with appropriate therapy
- Aldosterone "escape" justifies MRA (spironolactone/eplerenone) even with ACE-I or ARB use
- Right HF most commonly results from left HF - not primary right-sided disease
Sources: Robbins, Cotran & Kumar - Pathologic Basis of Disease | Textbook of Family Medicine 9e | Goodman & Gilman's Pharmacological Basis of Therapeutics
What are the pharmacological treatments and their mechanisms in HF?
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive data on all major drug classes. Let me compile the full, organized answer.
Pharmacological Treatments in Heart Failure
The modern approach to HFrEF is built around four pillars that reduce neurohormonal overactivation and improve survival, plus symptom-relieving agents. HFpEF management is less evidence-based but overlapping.
The "Fantastic Four" - Disease-Modifying Drugs for HFrEF (Mortality Benefit)
These four classes are now recommended to be started as early as possible, ideally simultaneously or rapidly sequenced:
1. ARNI (Angiotensin Receptor-Neprilysin Inhibitor)
Drug: Sacubitril/valsartan (Entresto)
Mechanism:
- Sacubitril inhibits neprilysin - the enzyme that degrades natriuretic peptides (ANP, BNP), bradykinin, and adrenomedullin. This increases circulating natriuretic peptides → vasodilation, natriuresis, anti-fibrotic, anti-hypertrophic effects.
- Valsartan (ARB) blocks AT₁ receptors → reduces vasoconstriction, aldosterone secretion, and adverse remodeling.
- The ARB component is used (not ACE-I) because both neprilysin and ACE-I break down bradykinin; combining them risks dangerous angioedema.
Key trial: PARADIGM-HF (2014) - sacubitril/valsartan reduced cardiovascular mortality by ~20% vs. enalapril.
Important rules:
- Never combine with ACE inhibitor
- Require a 36-hour washout after stopping ACE-I before starting
- Contraindicated if history of angioedema
Note: BNP is elevated by sacubitril (not degraded), but NT-proBNP is unaffected - use NT-proBNP to monitor.
2. Beta-Blockers
Drugs (with proven mortality benefit): Carvedilol, bisoprolol, metoprolol succinate (CR/XL)
Mechanism:
- HF is characterized by chronic SNS hyperactivation: excess norepinephrine causes myocyte hypertrophy, apoptosis, Ca²⁺ overload, arrhythmias, increased afterload, and receptor downregulation.
- Beta-blockers counteract these effects: reduce HR (an independent prognostic factor), decrease myocardial O₂ demand, prevent arrhythmias, and allow adrenergic receptor upregulation over time.
- Long-term use improves EF (reverse remodeling) - paradoxically, despite initially reducing contractility.
Key trials: MERIT-HF (metoprolol, ~34% mortality reduction), COPERNICUS (carvedilol in severe HF).
Practical: Start LOW, titrate SLOW. Initiation during acute decompensation is not recommended. Do NOT abruptly stop.
Contraindications: Significant bradycardia, high-degree AV block, asthma (COPD is not an absolute contraindication - use β₁-selective agents).
3. MRA - Mineralocorticoid Receptor Antagonists
Drugs: Spironolactone, eplerenone, finerenone (newer, nonsteroidal)
Mechanism:
- Aldosterone promotes Na⁺/water retention, K⁺/Mg²⁺ loss, sympathetic activation, parasympathetic inhibition, myocardial and vascular fibrosis, and baroreceptor dysfunction - all deleterious in HF.
- Aldosterone "escapes" ACE-I/ARB therapy (chymase can still generate angiotensin II, and aldosterone is also regulated by plasma Na⁺/K⁺ levels) → MRA blockade is needed on top of RAAS blockade.
- MRAs are K⁺-sparing diuretics at the aldosterone-sensitive distal tubule/collecting duct, but their main HF benefit is anti-fibrotic and anti-remodeling.
Key trials: RALES (spironolactone ~30% mortality reduction, NYHA III-IV), EMPHASIS-HF (eplerenone).
Spironolactone vs. Eplerenone:
| Feature | Spironolactone | Eplerenone |
|---|---|---|
| Selectivity | Non-selective (also blocks androgen/progesterone receptors) | Selective mineralocorticoid |
| Gynecomastia | Yes (~10% men) | No |
| Hyperkalemia risk | Yes | Yes (slightly less) |
Finerenone (2021 FDA approval) - non-steroidal, more selective, may cause less hyperkalemia.
Major risk: Hyperkalemia. Monitor K⁺ and eGFR closely. Do not use if K⁺ >5.0 or eGFR <30.
4. SGLT2 Inhibitors
Drugs: Dapagliflozin, empagliflozin
Mechanism:
- Inhibit sodium-glucose co-transporter 2 (SGLT2) in the renal proximal tubule → increased urinary glucose AND sodium excretion + osmotic diuresis.
- Additional effects: increased hematocrit, reduced long-term GFR decline, modest BP reduction.
- Exact cardioprotective mechanism in HF is not fully established but extends beyond glycemic control - effective regardless of diabetes status.
- Mitigate MRA-induced hyperkalemia.
Key trials: DAPA-HF (dapagliflozin, ~17% mortality reduction in HFrEF regardless of DM), EMPEROR-Reduced (empagliflozin).
Advantages over other drug classes: Single fixed dose, no titration required. Beneficial in CKD as well.
Contraindications: Type 1 DM, history of DKA, eGFR <20 mL/min/1.73m².
Symptom-Relieving Drugs (Do NOT reduce mortality alone)
5. Diuretics
Drugs:
- Loop diuretics: Furosemide, torsemide, bumetanide (most potent)
- Thiazides: Metolazone (used synergistically in resistant oedema)
Mechanism:
- Loop diuretics: Inhibit the Na⁺-K⁺-2Cl⁻ symporter in the thick ascending limb of the loop of Henle → block up to 15% of filtrate reabsorption → powerful natriuresis and diuresis.
- Thiazides: Inhibit Na⁺-Cl⁻ co-transporter in the distal convoluted tubule.
- Move patients to lower filling pressures along the ventricular function curve - relieves congestion.
- Do NOT give to patients without congestion (activates RAAS, worsens neurohormonal state).
Key point: Diuretics are essential for symptoms but have no proven mortality benefit on their own. Diuretic resistance can occur in severe HF (use thiazide combination: loop + metolazone - monitor electrolytes carefully).
Second-Line / Adjunctive Agents
6. Hydralazine + Isosorbide Dinitrate (H-ISDN)
Mechanism:
- Hydralazine: Arteriolar vasodilator → reduces afterload
- Isosorbide dinitrate: Venodilator → reduces preload
- Together they reduce both pre- and afterload and may inhibit oxidative stress.
- Use: When ACE-I/ARB/ARNI are not tolerated (e.g., renal failure, hypotension, angioedema). Also specifically recommended for Black patients with HFrEF already on standard therapy (A-HeFT trial showed additive mortality benefit in this population).
7. Ivabradine
Mechanism:
- Selectively blocks the If (funny) current (HCN channels) in the SA node → reduces heart rate without affecting myocardial contractility or blood pressure.
- Used when: resting HR ≥70 bpm in sinus rhythm despite maximal tolerated beta-blocker dose (or where beta-blockers are contraindicated).
8. Vericiguat
Mechanism:
- Stimulates soluble guanylate cyclase (sGC) → increases cGMP → activates protein kinase G → vascular and cardiac muscle relaxation.
- Also inhibits pathological fibrosis and hypertrophy.
- For patients who remain symptomatic after recent hospitalization for HF worsening, despite full neurohormonal blockade.
- Reduces HF hospitalization but not all-cause mortality.
- Avoid with PDE5 inhibitors (risk of severe hypotension).
9. Digoxin
Mechanism:
- Inhibits Na⁺/K⁺-ATPase → increases intracellular Na⁺ → reduces Na⁺-Ca²⁺ exchanger activity → increases intracellular Ca²⁺ → positive inotropy.
- Also enhances parasympathetic tone and inhibits renin release.
- In AF with HF: rate control benefit.
- DIG trial: reduced hospitalizations but no mortality benefit.
- Narrow therapeutic window. Risk of toxicity (bradycardia, heart block, nausea, visual disturbances). Target digoxin level: 0.5-0.9 ng/mL.
Summary Table
| Drug Class | Examples | Mechanism | Mortality Benefit |
|---|---|---|---|
| ARNI | Sacubitril/valsartan | Neprilysin inhibition + AT₁ block | ✅ Yes |
| Beta-blockers | Carvedilol, bisoprolol, metoprolol | Block SNS activation | ✅ Yes |
| MRA | Spironolactone, eplerenone | Block aldosterone receptor | ✅ Yes |
| SGLT2i | Dapagliflozin, empagliflozin | SGLT2 inhibition → natriuresis | ✅ Yes |
| Loop diuretics | Furosemide | Na⁺-K⁺-2Cl⁻ block (loop of Henle) | ❌ No (symptom relief) |
| Hydralazine/ISDN | - | Afterload + preload reduction | ✅ Selected patients |
| Ivabradine | - | If-current block in SA node | Partial (HR reduction) |
| Vericiguat | - | sGC stimulation → ↑cGMP | Partial (hospitalizations) |
| Digoxin | - | Na⁺/K⁺-ATPase inhibition | ❌ No |
High-Yield Exam Points
- ARNI + beta-blocker + MRA + SGLT2i = the "four pillars" of HFrEF - all recommended together
- ARNI preferred over ACE-I/ARB as first-line neurohormonal blockade
- ACE-I + neprilysin inhibitor is FORBIDDEN (angioedema risk) - 36 h washout required when switching ACE-I → ARNI
- Aldosterone escape justifies MRA despite already using ACE-I/ARB
- Spironolactone gynecomastia → switch to eplerenone
- SGLT2i benefit is DM-independent in HFrEF
- Digoxin - no mortality benefit; useful for symptom control and rate control in AF
- Beta-blockers improve EF over time (reverse remodeling) - don't judge beta-blocker efficacy by short-term contractility changes
- Do not start beta-blockers during acute decompensation
- Hydralazine/ISDN - specifically beneficial in Black patients and those intolerant of RAAS blockers
Sources: Goldman-Cecil Medicine | Goodman & Gilman's Pharmacological Basis of Therapeutics | Textbook of Family Medicine 9e
This is a shared conversation. Sign in to Orris to start your own chat.