Tumors like lesions of bone
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Now I have everything needed for a comprehensive answer. Here is the full response:
Tumor-Like Lesions of Bone
These are non-neoplastic bone lesions that mimic true bone tumors clinically and radiographically. They are classified under "Lesions Simulating Primary Neoplasms" in pathology. The major categories are covered below, along with key primary bone tumors for context.
Source: Robbins & Kumar Basic Pathology (Robbins Pathology)
Classification of Primary Bone Tumors (Overview)
| Category | Behavior | Tumor Type | Common Location | Age (yr) |
|---|---|---|---|---|
| Bone forming | Benign | Osteoid osteoma | Metaphysis of long bones | 10-20 |
| Bone forming | Benign | Osteoblastoma | Vertebral column | 10-20 |
| Bone forming | Malignant | Osteosarcoma | Distal femur, proximal tibia | 10-20 |
| Cartilage forming | Benign | Osteochondroma | Metaphysis of long bones | 10-30 |
| Cartilage forming | Benign | Chondroma | Small bones of hands/feet | 30-50 |
| Cartilage forming | Malignant | Chondrosarcoma | Pelvis, shoulder | 40-60 |
| Unknown origin | Benign | Giant cell tumor | Epiphysis of long bones | 20-40 |
| Unknown origin | Benign | Aneurysmal bone cyst | Proximal tibia, distal femur, vertebra | 10-20 |
| Unknown origin | Malignant | Ewing sarcoma | Diaphysis of long bones | 10-20 |
True Tumor-Like Lesions (Lesions Simulating Primary Neoplasms)
These are the three classic tumor-like (non-neoplastic) lesions:
1. Fibrous Cortical Defect and Nonossifying Fibroma
- Nature: Common developmental abnormality - fibrous connective tissue replaces bone
- Incidence: Present in up to 50% of children older than 2 years
- Location: Eccentrically in the metaphysis of the distal femur and proximal tibia; almost half are bilateral or multiple
- Size distinction: <0.5 cm = fibrous cortical defect; those that grow to 5-6 cm = nonossifying fibroma
- Radiology: Sharply demarcated radiolucent masses surrounded by a thin rim of sclerosis - appearance is specific enough that biopsy is rarely necessary
Morphology: Gray to yellow-brown cellular lesions with cytologically bland fibroblasts arranged in a storiform (pinwheel) pattern, with macrophages that may be foamy or form multinucleate giant cells. Hemosiderin is commonly present.
Clinical: Most small lesions resolve spontaneously. Progressive enlargement may cause pathologic fracture. Treatment is curettage with possible bone grafting.
2. Fibrous Dysplasia
- Nature: Benign lesion resulting from a localized developmental arrest - all bone components are present but fail to differentiate into mature structures
- Genetics: Somatic gain-of-function mutations in GNAS1 (same gene mutated in pituitary adenomas) → constitutively active Gs-protein → increased cAMP → promotes cellular proliferation and disrupts osteoblast differentiation
- Phenotype depends on when during embryogenesis the mutation was acquired and what proportion of mesenchymal cells are affected
Forms:
- Monostotic - single bone involvement (most common)
- Polyostotic - multiple bones; may cause deformities and fractures into adulthood
- Mazabraud syndrome - fibrous dysplasia + soft tissue myxoma
- McCune-Albright syndrome - polyostotic fibrous dysplasia + café-au-lait skin pigmentations + endocrine abnormalities (especially precocious puberty)
Morphology: Intramedullary lytic lesions that may expand and cause bowing/cortical thinning. Periosteal reaction is usually absent. Composed of curvilinear trabeculae of woven bone without an osteoblast rim, surrounded by a moderately cellular fibroblastic proliferation. Cystic degeneration, hemorrhage, and foamy macrophages are common.
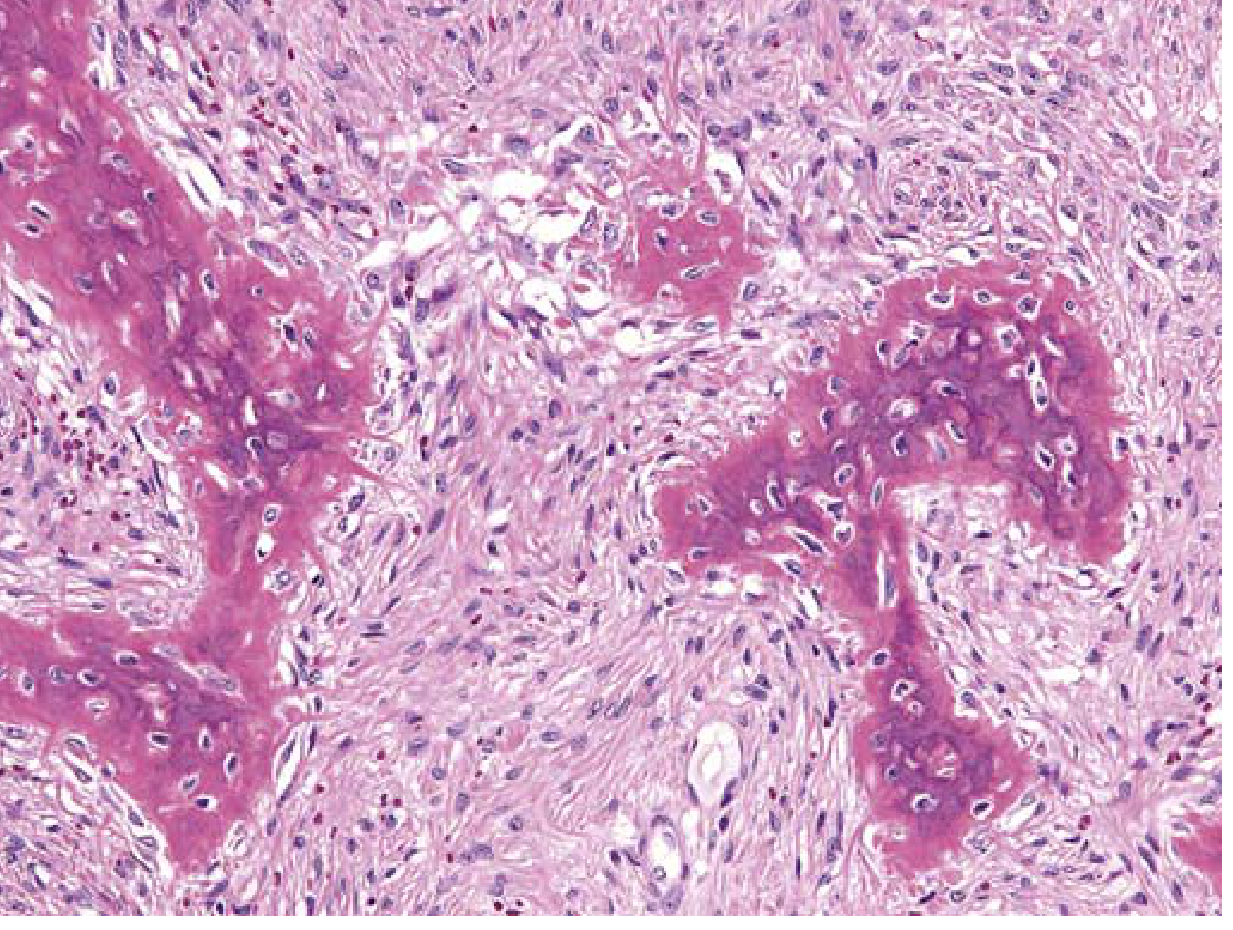
Key distinguishing feature: Absence of osteoblastic rimming of trabeculae (the "Chinese letters" pattern of woven bone in a fibrous stroma - likened to "alphabet soup")
Clinical:
- Monostotic disease often stops enlarging at growth plate closure
- Symptomatic lesions treated by curettage (recurrence common)
- Bisphosphonates reduce bone pain in polyostotic disease
- Rare complication: malignant transformation into sarcoma (usually polyostotic)
3. Aneurysmal Bone Cyst (ABC)
- Nature: Characterized by multiloculated blood-filled cystic spaces - not a true cyst and not truly aneurysmal
- Age: All age groups affected; most cases present in adolescence
- Location: Most frequently the femur, tibia, and vertebral body posterior elements
- Genetics: Spindle-shaped cells frequently demonstrate rearrangements of chromosome 17p13 → fusion of USP6 gene (encodes a deubiquitinating enzyme) regulatory elements to COL1A1 → USP6 overexpression → upregulates NF-κB → increases matrix metalloproteases → cystic bone resorption
Morphology:
- Radiography: Eccentric, expansile, lytic, metaphyseal lesion with well-defined margins
- CT/MRI: Internal septa and characteristic fluid-fluid levels (pathognomonic)
- Gross: Multiple blood-filled cystic spaces separated by thin tan-white septa
- Micro: Septa lack endothelial lining and are composed of plump uniform fibroblasts, multinucleate osteoclast-like giant cells, and reactive woven bone
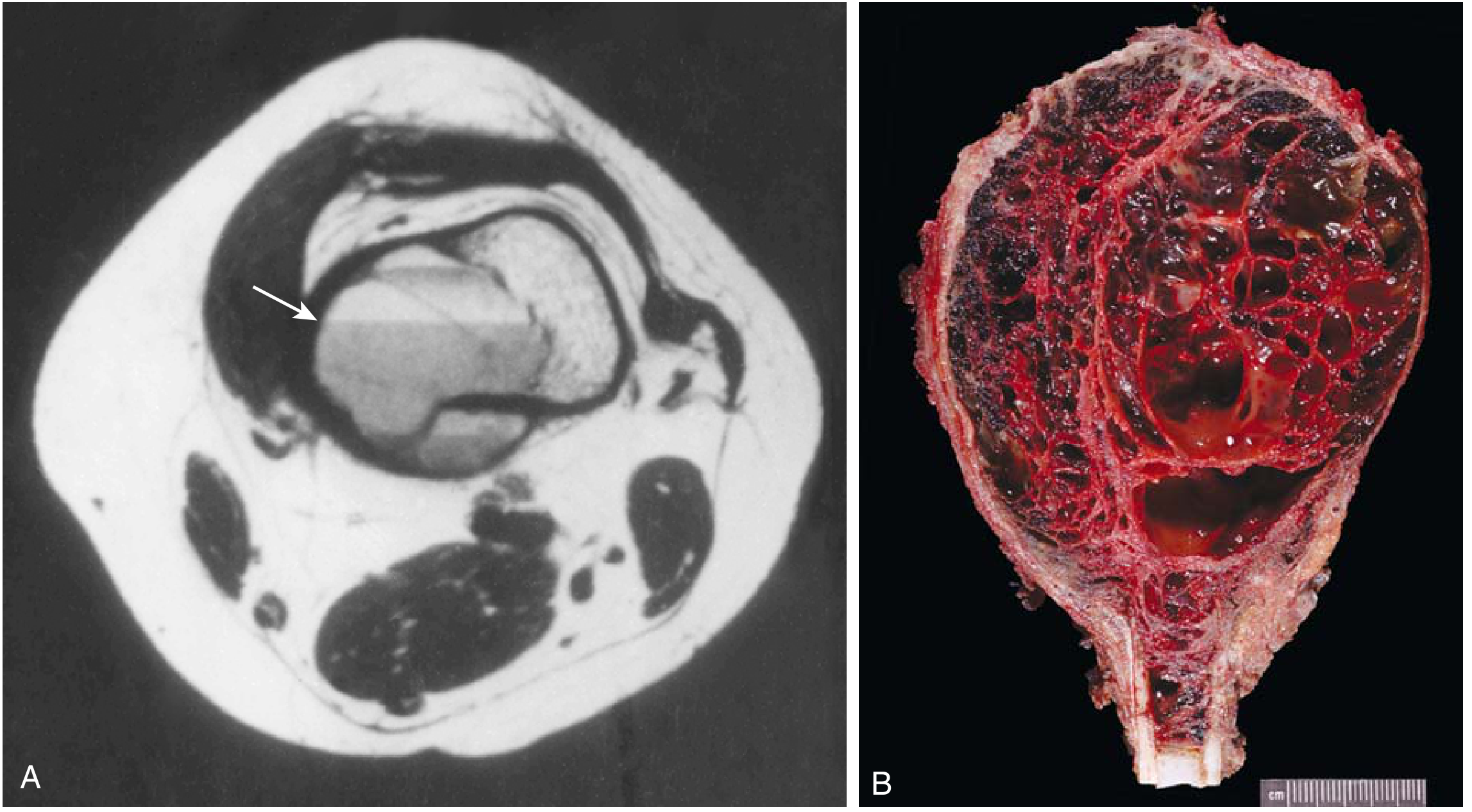
Clinical: Presents with localized pain and swelling. Although benign, it is locally aggressive. Treatment: curettage or excision. Recurrence in 10-50% of cases.
Related: Giant Cell Tumor (True Tumor Often Grouped Here)
Although a true neoplasm, Giant Cell Tumor (GCT) is often discussed alongside tumor-like lesions:
- Age: 20-40 years (adults after skeletal maturity)
- Location: Epiphysis of long bones (distal femur, proximal tibia - around the knee)
- Behavior: Locally aggressive, can rarely metastasize
- Genetics: H3F3A mutations (histone H3.3) in stromal cells; RANKL-dependent osteoclast recruitment
- Radiology: Lytic, expansile, destroys medullary canal and cortex; extends to subchondral bone
- Morphology: Sheets of uniformly distributed multinucleate osteoclast-like giant cells (up to 50+ nuclei) with background mononuclear stromal cells
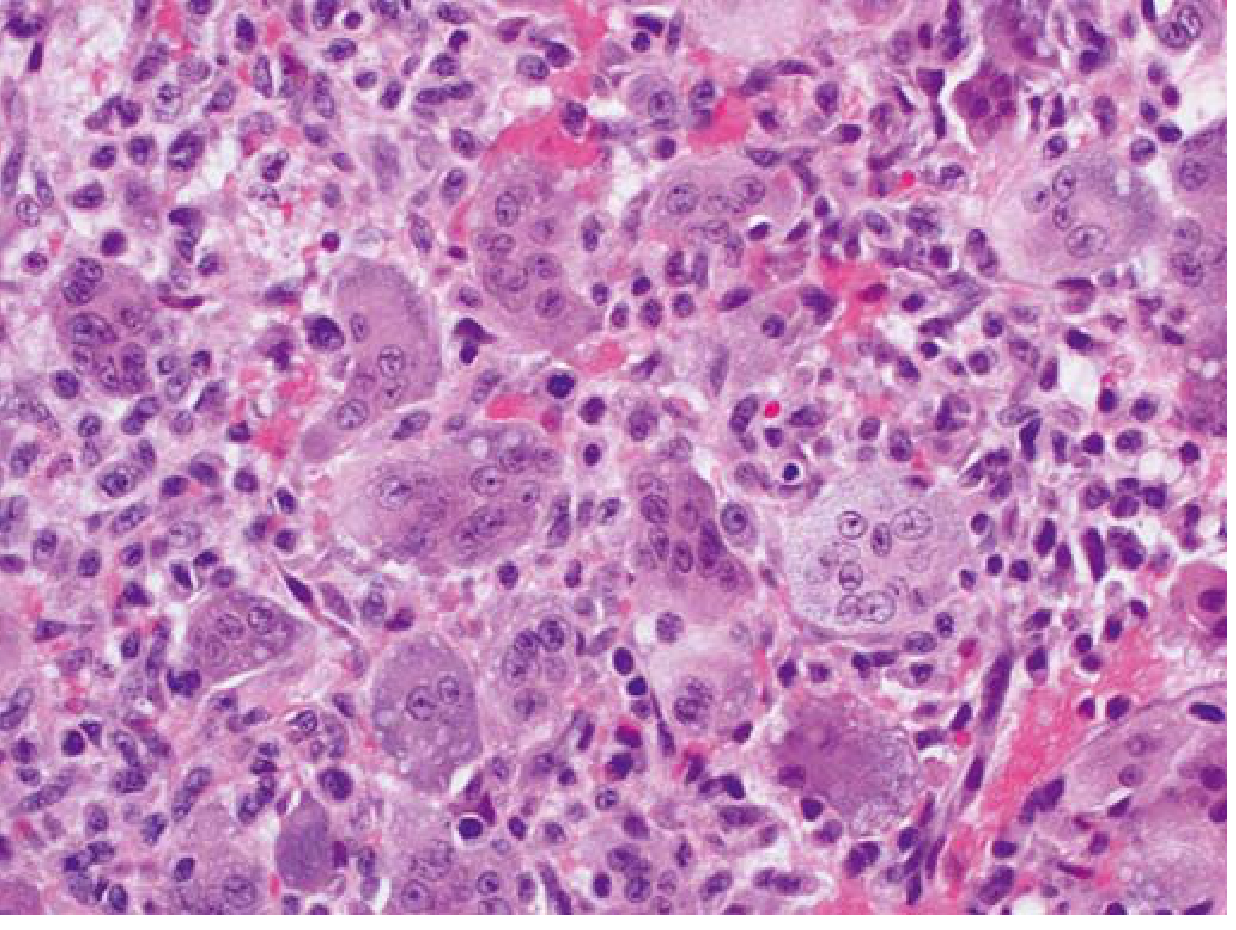
Treatment: Curettage with bone cement/grafting; denosumab (anti-RANKL) for unresectable cases. Recurrence ~50% after curettage alone.
Quick Comparison Summary
| Lesion | Age | Location | Key Feature | Radiology | Genetics |
|---|---|---|---|---|---|
| Fibrous cortical defect / NOF | Children/teens | Metaphysis of distal femur/tibia | Storiform fibroblasts, hemosiderin | Radiolucent with sclerotic rim | - |
| Fibrous dysplasia | Any (presents young) | Variable | Woven bone without osteoblast rimming | Lytic, bowing, "ground glass" | GNAS1 mutation |
| Aneurysmal bone cyst | Adolescents | Femur, tibia, vertebra | Blood-filled cysts, no endothelial lining | Eccentric lytic + fluid-fluid levels | USP6-COL1A1 fusion |
| Giant cell tumor | 20-40 | Epiphysis | Sheets of giant cells | Lytic, epiphyseal | H3F3A mutation |
- Robbins & Kumar Basic Pathology, pp. 780-789
This is a shared conversation. Sign in to Orris to start your own chat.