Arsenic poisioning
Reading File
Finding Sources
Finding Sources
Searching PubMed
arsenic poisoning toxicology
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive information. Let me compile the full answer.
Arsenic Poisoning
Forms and Sources of Arsenic
Arsenic occurs in three oxidation states: elemental, trivalent (arsenite), and pentavalent (arsenate). Common sources include:
- Inorganic compounds: Arsenic trioxide, Paris green (copper arsenite), calcium arsenate, lead arsenate - used as insecticides/pesticides
- Arsine gas (AsH3): Produced industrially when acid or water acts on arsenic-bearing metals
- Organic arsenicals: Herbicides, wood preservatives; less toxic than inorganic forms
- Environmental: Contaminated drinking water (permissible limit 0.05 mg/L)
- Occupational: Silicon chip manufacturing, smelters, agricultural workers
Pharmacokinetics
Absorption: Via oral ingestion, inhalation, dermal contact, or parenteral routes. On absorption, arsenic binds to the protein portion of hemoglobin.
Distribution: Arsenic distributes to virtually all tissues. Early on, the highest concentrations are in the liver, then kidneys and spleen. It does not cross the blood-brain barrier well (brain has lowest levels), but inorganic arsenic does cross the placenta. In prolonged exposure, arsenic deposits in keratin-rich tissues - hair, nails, and skin - for years, and replaces phosphorus in bone.
Elimination: Primarily via kidneys as methylated arsenic; also in feces, bile, and sweat. Detectable in urine within 30 minutes of ingestion, with continuous excretion for 10-12 days. Normal urine level is <0.03 µg/L.
Mechanism of Toxicity
Arsenic's toxicity operates through multiple pathways:
- Reversible combination with sulfhydryl (–SH) groups in enzymes and tissue proteins - this is the primary mechanism
- Interference with cellular metabolism - blocks multiple enzyme systems
- Capillary toxicity - dilates capillaries, causing third spacing and fluid loss
- Fatty degeneration of the liver
- Renal tubular necrosis
- Peripheral nerve damage - disintegration of axon cylinders (axonal neuropathy) with myelin fragmentation
Fatal Dose and Period
| Parameter | Value |
|---|---|
| Fatal dose (arsenic trioxide) | 180 mg (as low as 30 mg has been fatal) |
| Fatal period | 12-48 hours (can be fatal within 2-3 hours) |
Clinical Features
Acute Arsenic Poisoning
Symptoms typically begin within 30 minutes of ingestion:
| Stage | Features |
|---|---|
| Initial | Metallic taste, garlicky odour in breath, xerostomia (dry mouth), dysphagia |
| GI (cardinal) | Severe nausea & vomiting, colicky abdominal pain, profuse diarrhoea - initially bloody, later rice-water stools (resembling cholera), tenesmus |
| Cardiovascular | Third-spacing with shock, sinus/ventricular tachycardia, prolonged QT interval, ST depression, T-wave inversion, torsades de pointes, pericarditis |
| Respiratory | Pneumonia, pulmonary edema, ARDS, respiratory failure |
| Renal | Proteinuria, hematuria, oliguria, renal failure |
| Neurologic | Headache, drowsiness, delirium, coma, encephalopathy, seizures |
| Hepatic | Jaundice, pancreatitis, hepatomegaly |
(Source: ROSEN's Emergency Medicine, 10th ed.)
Differentiating Acute Arsenic Poisoning from Cholera
| Trait | Arsenic Poisoning | Cholera |
|---|---|---|
| Pain in throat | Before vomiting | After vomiting |
| Purging | After vomiting | Before vomiting |
| Stools | Dark/bloody, later rice-watery | Rice-watery, not bloody, involuntary jet |
| Tenesmus | Present | Absent |
| Vomited matter | Mucus, bile, blood | Watery, without mucus/bile/blood |
| Voice | Not affected | Rough and whistling |
| Conjunctivae | Inflamed | Not inflamed |
(Source: Essentials of Forensic Medicine & Toxicology, 36th ed., 2026)
Chronic Arsenic Poisoning
Neuropathy is the hallmark - symmetrical sensorimotor polyneuropathy, often resembling Guillain-Barre syndrome, with "glove and stocking" distribution, paresthesia, numbness, and pain especially on the soles. Can progress to muscular atrophy, wrist drop, inability to walk, and ataxia.
Skin changes (appear 1-4 weeks after acute episode):
- Early: erythematous flushing (capillary dilation)
- Aldrich-Mees lines (transverse white lines in fingernails) - appear ~5 weeks post-exposure; width ≥1-2 mm
- Raindrop pigmentation - finely mottled brown pigmentation on flexures, temples, eyelids, and neck
- Hyperkeratosis of palms and soles
- Diffuse, scaly desquamation with hyperpigmentation
- Brittle nails, patchy alopecia
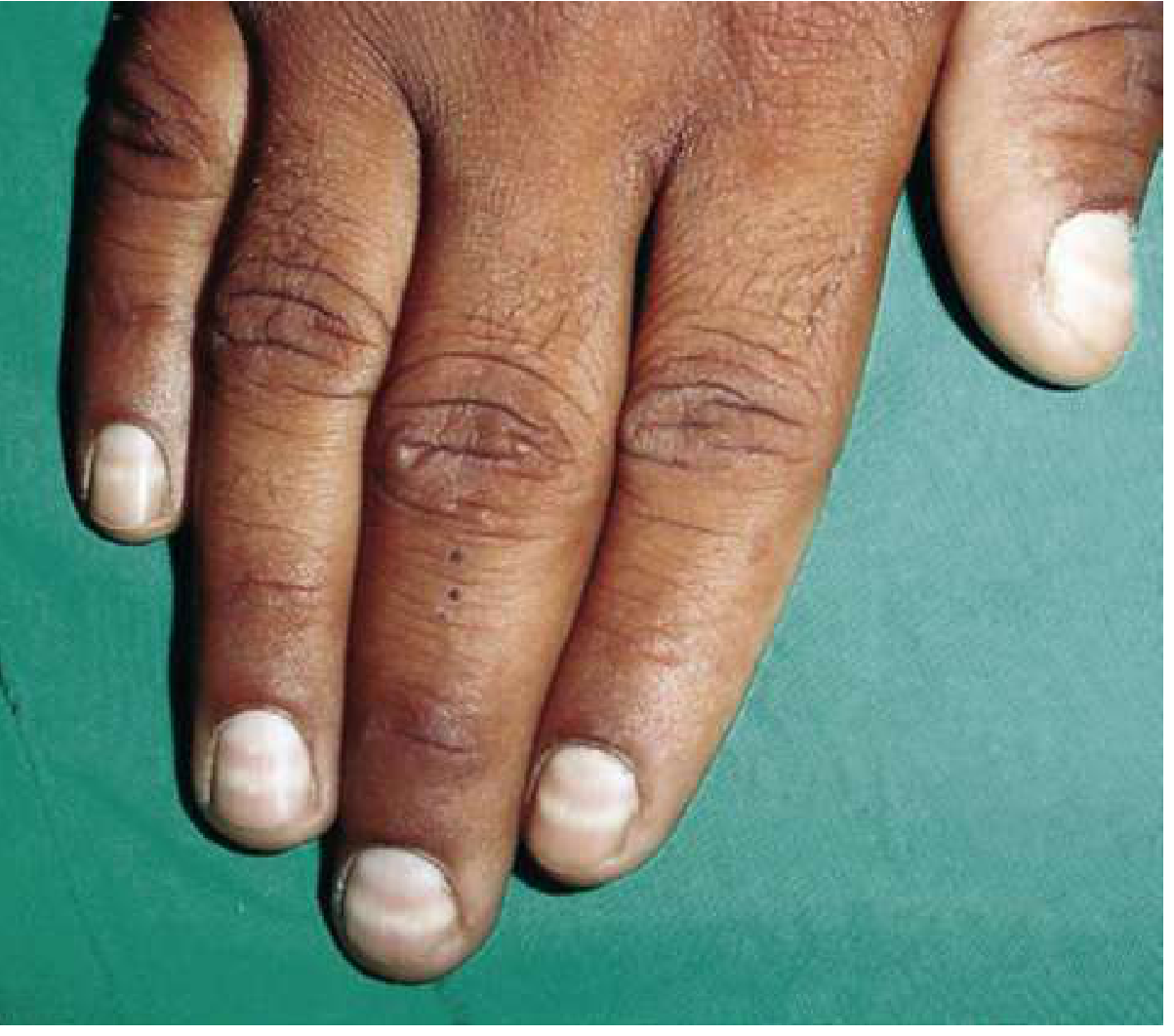
Other systemic features:
| System | Features |
|---|---|
| CNS | Encephalopathy, personality disturbance, convulsions, coma |
| Eyes | Conjunctivitis, photophobia, watering |
| GI | Nausea, vomiting, diarrhea, salivation |
| Hepatic | Hepatomegaly, jaundice, cirrhosis |
| Hematologic | Normochromic normocytic anemia (hemolysis), leukopenia, thrombocytopenia, mild eosinophilia, megaloblastic picture (folate interference), Bowen's disease |
| Renal | Chronic nephritis |
| CVS | Cardiac failure, dependent edema |
| Long-term | Lung cancer, skin cancer, bladder/kidney/liver cancer, leukemia; arsenic is teratogenic |
Arsine Gas Poisoning (Special Form)
Arsine gas causes severe acute hemolysis leading to renal tubular injury. Signs appear within minutes to hours. GI symptoms are common; CNS and hepatic dysfunction can occur. BAL is not effective for arsine gas poisoning.
Diagnosis
Definitive diagnosis is difficult because:
- Trace arsenic is naturally present in the body
- Multisystem toxicity mimics many diseases
Diagnostic tests:
| Sample | Threshold | Notes |
|---|---|---|
| 24-hr urine | >100 µg/day (acute) or >50 µg/L | Gold standard - spot urine is inaccurate |
| Urine (methylated metabolites) | >200 mg/24 hr | Monomethylarsine/dimethylarsine appear in urine within 24 hours |
| Blood | Normal: 100 µg/L | Useful only very soon after acute ingestion |
| Hair | >3 ppm (or >1 mg/100 g) | Useful for chronic/remote exposure; persists for years |
| Nails | >3 ppm or >100 µg/100 g | Same as hair |
Important caveat: Patients must avoid seafood (especially shellfish) before testing - organic arsenic (arsenobetaine) from seafood does NOT cause toxicity but falsely elevates total urine arsenic. Labs should specify the type of arsenic measured.
Other lab clues: Anemia, leukocytosis/leukopenia, erythrocyte basophilic stippling, elevated LFTs (AST/ALT/bilirubin), proteinuria, hematuria, prolonged QTc on ECG.
Radiograph: Arsenic is radiopaque in the GI tract (limited utility due to rapid absorption).
Detection methods: Atomic absorption spectroscopy, neutron activation analysis, colorimetry, polarography.
Historical note: Napoleon Bonaparte's death was attributed to chronic arsenic poisoning when hair analysis was performed 140 years after his death.
Treatment
Immediate / General
- Stabilization: Manage shock (aggressive IV fluids), dysrhythmias (QTc prolongation/torsades), and seizures
- Gastric decontamination: Gastric lavage with freshly prepared ferric oxide suspension or 1% sodium thiosulphate (ferric arsenite formed is harmless and insoluble). Avoid copper sulphate (forms toxic copper arsenite). Activated charcoal does NOT adsorb arsenic - not recommended
- Cathartics: Castor oil or magnesium sulphate to reduce intestinal absorption
- IV saline for dehydration
Chelation Therapy (Antidotes)
| Agent | Route | Notes |
|---|---|---|
| BAL (British Anti-Lewisite / Dimercaprol) | IM | Preferred for critically ill patients; 2.5-3 mg/kg, 4-hrly x 2 days, 6-hrly x 1 day, then 12-hrly; stop if side effects appear (nausea, tachycardia, hypotension, throat constriction) |
| Succimer (DMSA) | Oral | Effective but often limited by severe GI symptoms from poisoning |
| D-Penicillamine | Oral | 100 mg/kg/day in 4 divided doses for 4-8 days after initial BAL; use only if BAL/succimer unavailable (high side-effect profile) |
- Monitor 24-hr urine arsenic during chelation; stop when levels fall below 50 µg/24 hr (pause 5 days, then restart to mobilize tissue stores)
- BAL is not effective for arsine gas poisoning
- Hemodialysis may remove some arsenic in acute renal failure
- Exchange transfusion or plasma exchange for severe arsine gas exposure
Autopsy Findings
- Prolonged rigor mortis
- General shrunken appearance (dehydration)
- Shrunken eyeballs
- Cyanosis of hands and feet; jaundice
- Inflamed, ulcerated mucous membranes of mouth and pharynx
- Stomach: main organ affected - mucosa congested, brownish-red to scarlet, petechial hemorrhages; greatest involvement at the greater curvature and cardiac end
- Small intestine: flabby, submucosal hemorrhages, oedematous epithelium
- Renal tubular necrosis
- Liver: fatty degeneration
Long-Term Carcinogenic Risk
Chronic arsenic exposure is associated with:
- Skin - squamous cell carcinoma, Bowen's disease (in-situ SCC)
- Bladder, kidney, liver, lung cancers
- Leukemia
- Cardiovascular disease and diabetes mellitus (via contaminated drinking water)
A recent meta-analysis (Wang et al., 2025) confirmed that arsenic exposure through drinking water is significantly associated with pigment disorders in a dose-dependent manner.
Sources:
- P C Dikshit Textbook of Forensic Medicine and Toxicology
- ROSEN's Emergency Medicine: Concepts and Clinical Practice, 10th ed.
- The Essentials of Forensic Medicine and Toxicology, 36th ed. (2026)
- Katzung's Basic and Clinical Pharmacology, 16th ed.
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Kaplan and Sadock's Synopsis of Psychiatry
Create a clinical reference guide on heavy metal poisonings
Say the answer in the previous manner Not as ug standards
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Why minamata diesease doesn't need chelation
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Excellent - all the key information is in hand. Here is the complete answer:
Why Minamata Disease Does NOT Respond to Chelation
What is Minamata Disease?
Minamata disease is chronic methylmercury (organic mercury) poisoning - named after the mass poisoning in Minamata Bay, Kyushu, Japan between 1953 and 1956, where villagers ingested fish contaminated with industrial waste containing methylmercury. The cardinal features were:
- Concentric constriction of visual fields
- Hearing loss
- Cerebellar ataxia
- Postural and action tremors
- Sensory impairment of limbs, tongue and lips
Pathologically, there was diffuse neuronal loss in both cerebral and cerebellar cortices, most marked in the calcarine cortex (visual) and granule cell layer of the cerebellum.
The Core Reason: Chelation Makes It WORSE
Chelation does not just fail in methylmercury poisoning - in the case of BAL (dimercaprol), it is actively contraindicated because it has been shown to increase the concentration of methylmercury in the brain.
This is the central problem, and it has three layers of explanation:
1. Methylmercury Has Unique Pharmacokinetics
| Property | Inorganic Mercury (HgCl2) | Methylmercury (organic) |
|---|---|---|
| Water solubility | High | Highly lipophilic |
| Blood-brain barrier crossing | Poor | Crosses easily - actively transported via amino acid carrier (mimics methionine) |
| Protein binding | Binds to plasma proteins | Binds to erythrocyte hemoglobin sulfhydryl groups (90% in RBCs) |
| Primary excretion route | Kidneys (urine) | Bile/feces - enterohepatic recirculation |
| Primary toxic target | Kidneys | CNS - irreversibly |
| Tissue redistribution by BAL | Removed from periphery | Redistributed INTO the brain |
Methylmercury is lipophilic and actively transported across the blood-brain barrier. Once inside neurons, it causes irreversible neuronal death - the damage is structural, not biochemical-reversible.
2. BAL Redistributes Methylmercury into the CNS
Dimercaprol (BAL) works by chelating metal ions in tissues and blood, pulling them into a water-soluble complex excreted in bile and urine. For inorganic mercury, this is beneficial. For methylmercury:
"Dimercaprol has been shown to redistribute mercury to the central nervous system from other tissue sites, and since the brain is a key target organ, dimercaprol should not be used in treatment of chronic exposure to elemental or organic mercury."
- Katzung's Basic and Clinical Pharmacology, 16th ed.
"In methylmercury and alkylmercury poisoning, dimercaprol is contraindicated because it has been found to increase the concentrations of these compounds in the brain."
- Henry's Clinical Diagnosis and Management by Laboratory Methods
So BAL actively worsens the neurological damage by driving more methylmercury across the blood-brain barrier.
3. The Damage is Neuronal Death - Not Enzyme Inhibition
Chelation therapy works when a metal is blocking an enzyme's sulfhydryl group - the chelator competitively displaces the metal and the enzyme function is restored. This is reversible.
In Minamata disease, methylmercury causes:
- Degeneration of the granular layer of the cerebellar cortex
- Neuronal loss and gliosis of the calcarine cortex
- Diffuse cortical neuronal death
Dead neurons cannot be "unchained" from a metal. The structural damage is already done. There is no enzymatic function to rescue - the cells are gone.
4. Methylmercury Is Excreted via Bile, Not Urine
Chelation agents like EDTA or BAL work by forming metal-chelate complexes that are excreted in the urine. Methylmercury undergoes enterohepatic recirculation - it is excreted into bile, reabsorbed from the gut, and recirculated. A chelator that promotes renal excretion has limited impact on a toxin that is not primarily renally eliminated.
Short-chain alkyl organic mercury compounds are mainly excreted in the bile, so blood levels (not urine) are better indicators of tissue burden.
- Henry's Clinical Diagnosis and Management by Laboratory Methods
What IS Used (with Limited Evidence)
Since no chelation is effective, treatment in methylmercury poisoning is primarily supportive. Some agents have limited data:
| Agent | Mechanism | Evidence |
|---|---|---|
| Succimer (DMSA) | Oral chelator, may enhance body clearance | Limited data - may modestly increase methylmercury excretion |
| Unithiol (DMPS) | Similar to DMSA | May increase urine mercury excretion |
| N-acetyl-L-cysteine (NAC) | Sulfhydryl donor, may help gut excretion | Limited data |
| Activated charcoal / polystyrene resin | Interrupts enterohepatic recirculation in the gut | Most rational approach for acute ingestion |
"Limited data suggest that succimer, unithiol, and N-acetyl-L-cysteine (NAC) may enhance body clearance of methylmercury."
- Katzung's Basic and Clinical Pharmacology, 16th ed.
Summary
Chelation fails in Minamata disease for four converging reasons:
- BAL actively redistributes methylmercury INTO the brain - it is contraindicated, not just ineffective
- Methylmercury is lipophilic and crosses the BBB freely - tissue damage is at a neuronal level, not vascular/renal
- The neuronal death is irreversible - no chelator can restore dead neurons
- Methylmercury is excreted via bile (enterohepatic circulation), not urine - the typical chelation-then-renal-excretion mechanism does not apply
(Sources: Adams and Victor's Principles of Neurology 12th ed.; Katzung's Basic and Clinical Pharmacology 16th ed.; Henry's Clinical Diagnosis and Management by Laboratory Methods; Essentials of Forensic Medicine & Toxicology 36th ed., 2026)
How bone line formed in lead poisioning
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
All the information needed is gathered. Here is the complete answer:
How Lead Lines Form in Bone (Lead Poisoning)
The Key Concept First - A Common Misconception
The first thing to understand is that lead lines on X-ray are NOT simply lead deposited in bone. They represent failure of normal bone remodeling (hypermineralization) at the growth plate. The dense white band you see is calcified cartilage that accumulates because lead prevents it from being resorbed and replaced by normal bone.
Step-by-Step Mechanism of Formation
Step 1 - Lead Mimics Calcium in the Body
Lead is chemically similar to calcium (both are Group IIA/divalent cations). When lead is absorbed into the bloodstream, it is handled by the body as if it were calcium:
- It is drawn towards rapidly growing areas of the skeleton - specifically the metaphysis (the zone just below the growth plate where bone is actively forming)
- It is stored in bone as insoluble lead phosphate and lead carbonate (just as calcium is stored as calcium phosphate/hydroxyapatite)
- High calcium levels favour lead storage in bone; calcium deficiency causes lead to be released back into the bloodstream - this is why dietary calcium status matters clinically
Step 2 - Lead Concentrates at the Growth Plate (Physis)
Lead is drawn most strongly to the areas of fastest skeletal growth:
- Distal femur
- Proximal tibia
- Distal radius
The metaphysis is where the growth plate's calcified cartilage normally undergoes remodeling - osteoclasts resorb it and osteoblasts lay down lamellar bone in its place. This is a continuous, active process in growing children.
Step 3 - Lead Interferes with Normal Growth Plate Remodeling
Lead inhibits the normal remodeling of the growth plate (physis):
- In the normal growth plate, calcified cartilage in the zone of provisional calcification is continuously resorbed and replaced by trabecular bone
- Lead disrupts the osteoclast-mediated resorption of this calcified cartilage
- The result: calcified cartilage accumulates and is not resorbed - it piles up as an abnormally thick, dense zone at the metaphysis
- This is hypermineralization - excess mineralized tissue, not lead metal deposits
"Excess lead interferes with the normal remodeling of the growth plate (physis) in children, causing increased bone density detected as radio-dense 'lead lines'."
- Robbins & Kumar Basic Pathology
"Lead lines seen on X-rays as densities are due to hypermineralization. These lines reflect 'bone growth arrest'."
- Essentials of Forensic Medicine & Toxicology, 36th ed. (2026)
"Horizontal, metaphyseal 'lead lines' represent failure of bone remodeling rather than deposition of lead."
- Tintinalli's Emergency Medicine
Step 4 - The Radiodense Band Appears
Because the calcified cartilage zone has accumulated to an abnormal thickness, it becomes as radiopaque as cortical bone on X-ray - appearing as a brilliant white horizontal band across the metaphysis.
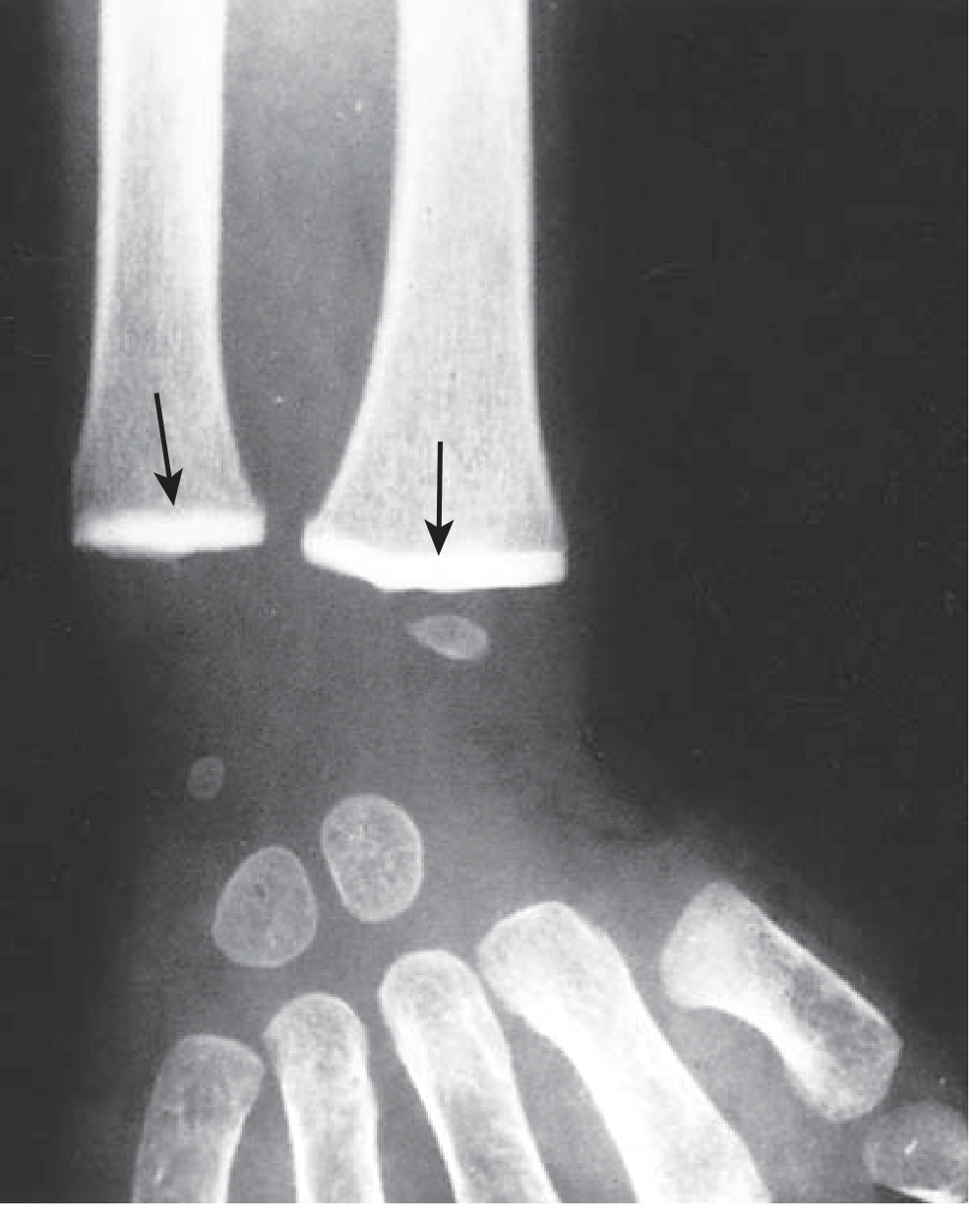
Key Features of Lead Lines in Bone
| Feature | Detail |
|---|---|
| Location | Metaphysis of long bones (distal femur, proximal tibia, distal radius, iliac crest) |
| Appearance | Dense transverse (horizontal) white bands across the metaphysis |
| Mechanism | Hypermineralization - failure of calcified cartilage resorption |
| Minimum duration | Seen only after at least 4 weeks of heavy chronic poisoning |
| Width | Proportional to duration of lead exposure |
| Multiple lines | Indicate repeated episodes of toxicity (like tree rings) |
| Age group | Most commonly seen between 2 to 5 years of age (peak bone growth) |
| Best bones to X-ray | Knee (distal femur + proximal tibia) gives the clearest lines |
Additional Effect: Lead Also Inhibits Fracture Healing
Lead increases chondrogenesis and delays cartilage mineralization in fracture repair - so fractures heal poorly in chronic lead poisoning, by a related but separate mechanism.
Why Children and Not Adults?
Lead lines are essentially a paediatric finding because:
- The growth plate (physis) is only active in children - it closes at skeletal maturity
- Adults have no active endochondral ossification to disrupt
- The metaphysis in adults is remodeled long bone - there is no "zone of provisional calcification" to accumulate
In adults, lead still deposits in bone but does not produce the characteristic radiodense line because there is no active growth plate remodeling to disrupt.
Summary in One Sentence
Lead lines form because lead concentrates at the metaphysis of growing bones, poisons the osteoclasts responsible for resorbing calcified cartilage at the growth plate, causing it to accumulate as an abnormally thick, hypermineralized band - visible as a dense white line on X-ray.
(Sources: Robbins & Kumar Basic Pathology; Essentials of Forensic Medicine & Toxicology 36th ed. 2026; Tintinalli's Emergency Medicine)
Gastric lavage in phosphorus poisioning
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.