How exactly tablet works in our body
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed for a thorough, visually-supported answer. Here's the complete explanation:
How a Tablet Works in Your Body
This field is called pharmacokinetics - literally "what the body does to a drug." There are four core stages, often abbreviated ADME.
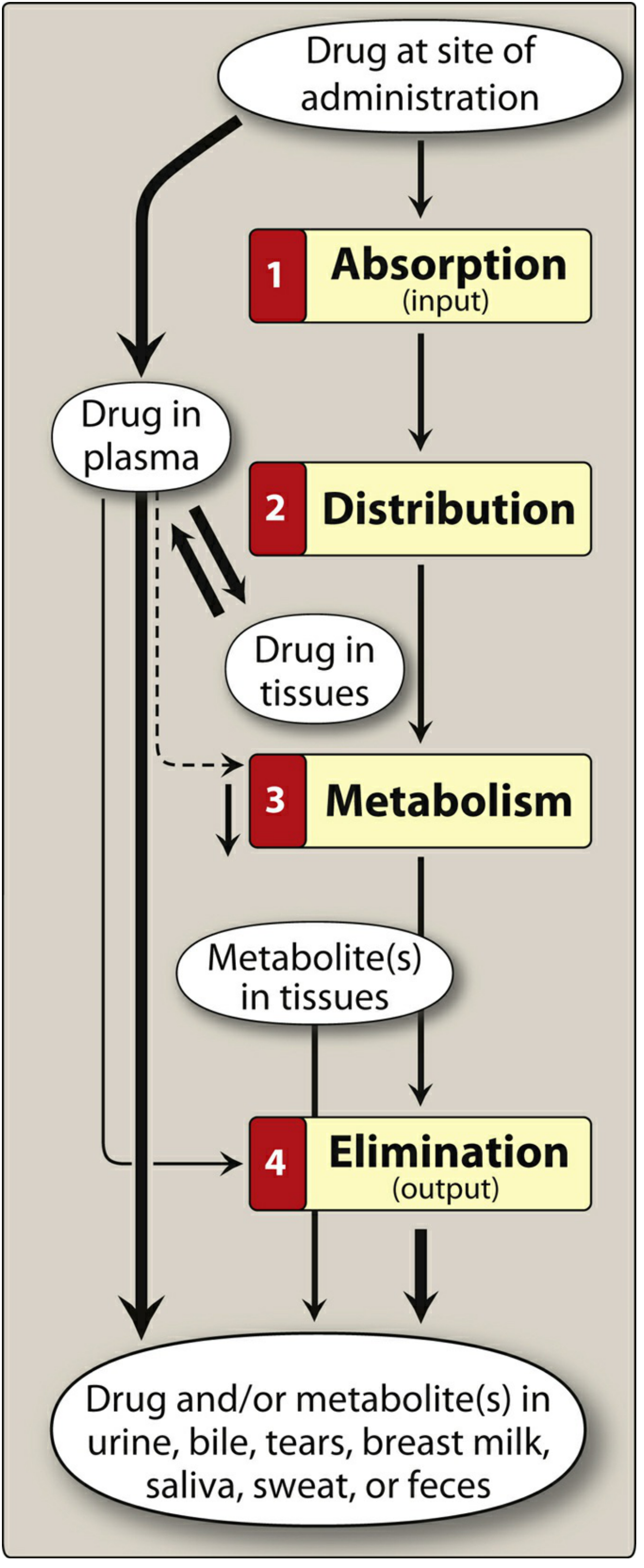
Stage 1 - Absorption
When you swallow a tablet, it travels to the stomach and small intestine. Three things happen:
- Disintegration - The tablet physically breaks apart into smaller granules in the stomach fluid.
- Dissolution - The drug dissolves into the gastric/intestinal fluid.
- Crossing the gut wall - The dissolved drug must cross the intestinal lining to enter the bloodstream. This is where lipid solubility matters enormously.
As Lippincott Illustrated Reviews: Pharmacology explains: drugs that are very hydrophilic (water-loving) are poorly absorbed because they cannot cross the lipid-rich cell membranes. Drugs that are extremely lipophilic (fat-loving) are also poorly absorbed because they are insoluble in the watery gut fluid. The ideal drug is mostly lipophilic, but slightly water-soluble - which is why many drugs are designed as weak acids or weak bases.
The small intestine is the primary absorption site because of its enormous surface area (villi and microvilli).
Stage 2 - First-Pass Metabolism (The Liver Filter)
This is the most misunderstood step. Before the drug reaches the rest of your body, it goes through what's called the portal circulation:
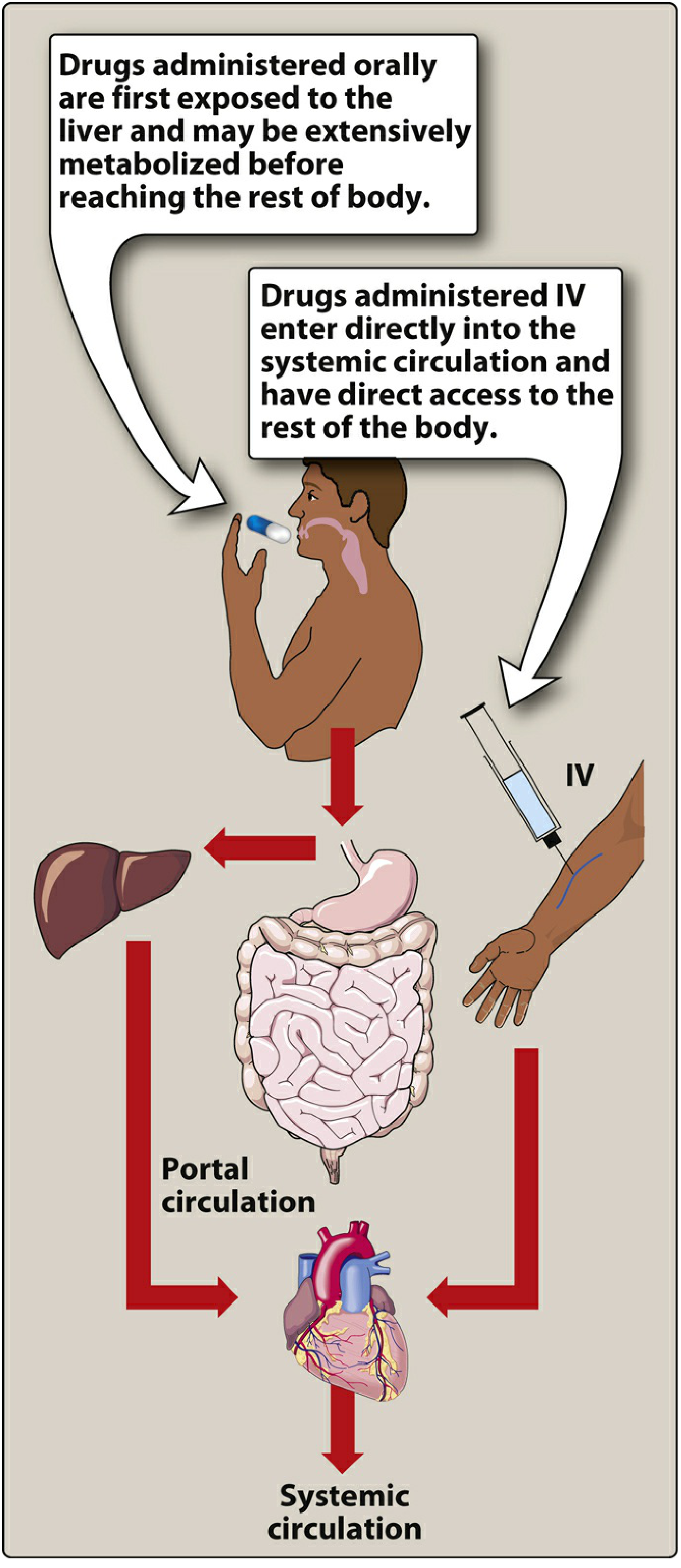
Blood from your gut drains into the portal vein, which goes directly to the liver - before reaching the heart or the rest of the body. The liver enzymes immediately begin breaking down (metabolizing) some of the drug. This is called first-pass metabolism or pre-systemic elimination.
For some drugs, this effect is enormous. For example, more than 90% of nitroglycerin is destroyed during first-pass metabolism - which is why it is given under the tongue (sublingual), bypassing the liver entirely. - Lippincott Pharmacology
Bioavailability is the term for how much of the drug actually makes it into your systemic circulation unchanged. If you take 100 mg of a drug with 70% bioavailability, only 70 mg reaches your bloodstream to exert its effect.
Stage 3 - Distribution
Once in the bloodstream, the drug is distributed throughout the body. It can:
- Stay in the plasma (blood fluid)
- Move into interstitial fluid (fluid around cells)
- Enter cells (intracellular fluid)
- Bind to plasma proteins like albumin (only unbound drug is active)
The volume of distribution (Vd) describes how widely the drug spreads. A drug with a large Vd is sequestered heavily in tissues (e.g., fat or muscle), while a low Vd drug stays mostly in blood.
Some drugs cannot easily enter the brain because of the blood-brain barrier - a tight, protective layer of cells. Drugs that need to act on the CNS must be lipophilic enough to cross it.
Stage 4 - Metabolism (Biotransformation)
The liver is the primary site of drug metabolism, using enzyme systems - chiefly the cytochrome P450 (CYP) enzymes. The liver converts the drug (usually a lipophilic molecule) into more polar (water-soluble) metabolites.
This happens in two phases:
- Phase I - Oxidation, reduction, or hydrolysis (usually by CYP enzymes) - adds or unmasks a reactive group
- Phase II - Conjugation (e.g., glucuronidation) - attaches a large polar molecule, making the drug water-soluble and ready for excretion
The metabolites are usually inactive, but sometimes a drug is given as an inactive "prodrug" that becomes active only after liver metabolism (e.g., codeine is converted to morphine).
Kinetics of elimination:
- Under first-order kinetics (most drugs): a constant fraction is eliminated per unit time. With each half-life (t½), the plasma concentration drops by 50%.
- Under zero-order kinetics (alcohol, aspirin at high doses): a constant amount is eliminated per unit time regardless of concentration.
Stage 5 - Excretion (Elimination)
The kidneys are the primary organ of excretion. They filter water-soluble metabolites from the blood into urine. Drugs and metabolites can also exit via:
- Bile/feces (biliary excretion)
- Exhaled air (e.g., volatile anesthetics, alcohol)
- Sweat, saliva, tears, breast milk (minor routes)
Summary Table
| Stage | Where | Key Concept |
|---|---|---|
| Disintegration & Dissolution | Stomach | Tablet breaks down, drug dissolves |
| Absorption | Small intestine | Drug crosses gut wall into portal blood |
| First-pass metabolism | Liver | Part of the drug is destroyed before reaching systemic circulation |
| Distribution | Blood + tissues | Drug spreads throughout body; Vd determines extent |
| Metabolism | Liver (mainly) | CYP enzymes convert drug to water-soluble metabolites |
| Excretion | Kidneys (mainly) | Metabolites filtered out in urine |
Why Timing Matters
Each drug has a half-life - the time it takes for plasma concentration to fall by half. This determines:
- How often you need to take it (dosing interval)
- How long it takes to leave your system
- How long to reach steady state (stable, effective blood levels) - typically after 4-5 half-lives
For example, a drug with a 6-hour half-life reaches steady state after about 24-30 hours of regular dosing.
Source: Lippincott Illustrated Reviews: Pharmacology; Goodman & Gilman's The Pharmacological Basis of Therapeutics
This is a shared conversation. Sign in to Orris to start your own chat.