Give me a comprehensive and detailed analysis on "Acute fatty liver in pregnancy"
Reading File
Finding Sources
Finding Sources
Searching PubMed
"acute fatty liver of pregnancy"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading Article
PMID: 35276220
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive data from multiple authoritative textbooks. Let me compile the full analysis.
Acute Fatty Liver of Pregnancy (AFLP): Comprehensive Analysis
Overview
Acute fatty liver of pregnancy (AFLP) is a rare but life-threatening obstetric emergency characterized by microvesicular fatty infiltration of hepatocytes leading to hepatic failure. It is unique to human gestation and presents predominantly in the third trimester. Originally considered almost universally fatal, modern outcomes with early diagnosis and prompt delivery are dramatically improved - maternal survival now approaches nearly 100% in well-resourced settings.
Epidemiology
-
Incidence: Approximately 1 in 6,700 to 1 in 16,000 pregnancies; a large UK national prospective cohort (1.1 million deliveries) found 5.0 cases per 100,000 pregnancies
-
Gestational age: Most commonly presents between 34-37 weeks, though cases as early as 19-20 weeks have been reported; rarely presents postpartum
-
Risk factors:
- Primiparity (first pregnancy)
- Multiple gestations (twin pregnancies independently identified as a risk factor)
- Low BMI (<20)
- Male fetus (affected patients have a significantly greater than expected proportion of male fetuses)
- Pre-existing fatty acid oxidation disorders in the fetus (LCHAD deficiency - see Pathogenesis)
-
Co-existing preeclampsia: Present in 21-64% of AFLP cases, reflecting shared pathophysiologic mechanisms and overlapping risk factors
-
Sleisenger and Fordtran's Gastrointestinal and Liver Disease, p. 661
-
Yamada's Textbook of Gastroenterology, p. 831
Pathogenesis
The pathogenesis of AFLP involves a fascinating fetal-maternal metabolic interaction.
LCHAD Deficiency and Beta-Oxidation Defects
The most established mechanism links AFLP to inherited defects in mitochondrial fatty acid beta-oxidation, particularly long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency:
- The fetus inherits two mutant copies of the HADHA gene (encoding LCHAD), making it homozygous deficient
- The mother and father are each obligate heterozygous carriers - insufficient for disease outside pregnancy
- The enzyme deficiency causes the fetus and placenta to spill unmetabolized long-chain 3-hydroxyacyl fatty acid metabolites into the maternal circulation
- These metabolites accumulate in the mother's liver, causing hepatotoxicity and the clinical syndrome of AFLP
- This represents one of the rare instances where the fetus causes metabolic disease in the mother
AFLP may develop regardless of maternal genotype if the fetus is LCHAD-deficient and carries at least one allele for the G1528C LCHAD mutation. Another beta-oxidation defect - carnitine palmitoyltransferase-1 deficiency - has also been associated with AFLP.
Empirical support comes from the clinical similarity between AFLP and Jamaican vomiting sickness, caused by hypoglycin A from unripe akee fruit, which directly inhibits intramitochondrial beta-oxidation.
Note: Not all AFLP cases are explained by LCHAD deficiency. An intensive search has failed to identify a single toxin responsible, and other unknown mechanisms likely exist.
Relationship to Preeclampsia
Because preeclampsia co-occurs in 21-64% of AFLP cases, and both share risk factors (primiparity, twins, male fetus), a subset of investigators consider AFLP a severe form of preeclamptic liver disease. Placental oxidative stress with release of toxic mediators may play a role. However, AFLP liver biopsies typically lack the classic histologic features of preeclampsia, arguing against this being the sole mechanism.
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. (pathogenesis section)
- Sleisenger and Fordtran's GI and Liver Disease, p. 661-662
- Comprehensive Clinical Nephrology, 7th Ed., p. 633
Pathology / Histology
The histologic hallmark of AFLP is diffuse microvesicular steatosis - small fat droplets within hepatocytes that do not displace the nucleus (unlike macrovesicular fat, which displaces the nucleus to the periphery).
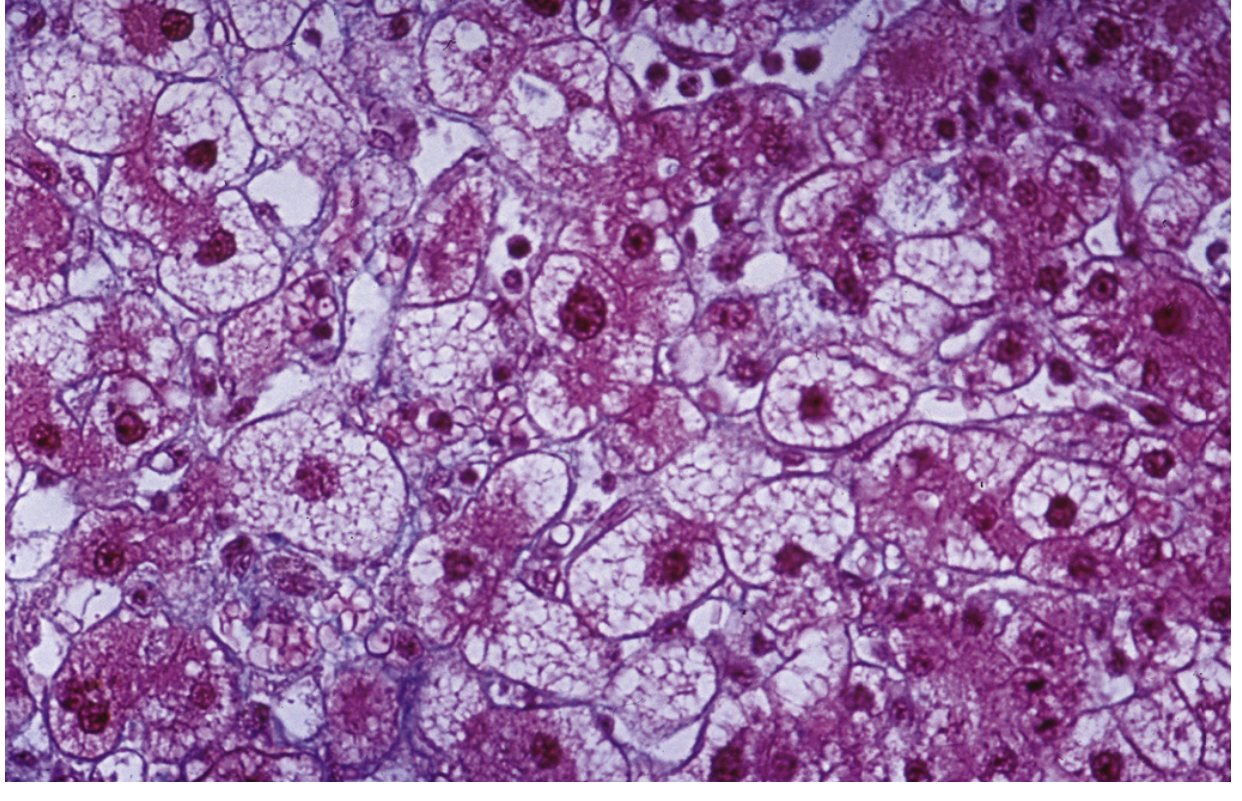
Histopathology of acute fatty liver of pregnancy. The perivenular hepatocytes are pleomorphic and vacuolated, and there is lobular disarray. Large fat droplets are not seen. (From Sleisenger and Fordtran's GI and Liver Disease)
Zonal Pattern
- Steatosis is most prominent in zone 3 hepatocytes (perivenular/centrilobular) and spares zone 1 hepatocytes surrounding portal areas
- The distribution is relatively homogeneous on light microscopy
Staining Challenges
- Microvesicular fat has a relatively homogeneous, pale appearance on H&E and may be difficult to discern
- Confirmation requires:
- Oil-Red O stain on frozen tissue (stains fat red-orange)
- Electron microscopy on glutaraldehyde-fixed specimen (shows characteristic mitochondrial pleomorphism)
Other Histologic Findings
-
Lobular disarray (may mimic viral hepatitis)
-
Hepatocyte dropout with reticulin collapse
-
Biliary ductular proliferation
-
Cholestasis may be present
-
Notable for: relative absence of inflammation and necrosis in early disease (distinguishing from viral hepatitis)
-
Sleisenger and Fordtran's GI and Liver Disease, p. 661
-
Robbins, Cotran & Kumar Pathologic Basis of Disease, p. (morphology)
Clinical Presentation
Timing
- Late pregnancy, usually 34-37 weeks (third trimester)
- Rarely before 20 weeks or postpartum
Prodromal Symptoms (Days to Weeks Before Diagnosis)
- Nausea and vomiting (most common)
- Abdominal pain (often right upper quadrant or epigastric)
- Malaise, anorexia
- Polydipsia/polyuria (may reflect early transient diabetes insipidus)
- Pruritus (overlap with intrahepatic cholestasis of pregnancy - rare)
Established Disease
| System | Manifestation |
|---|---|
| Hepatic | Jaundice, hepatic encephalopathy, coagulopathy, right upper quadrant pain |
| Metabolic | Hypoglycemia (hallmark of hepatic synthetic failure), hyperammonemia |
| Renal | Acute kidney injury - elevated creatinine, BUN, and uric acid |
| Hematologic | Leukocytosis, DIC, thrombocytopenia |
| CNS | Confusion, altered consciousness (from hypoglycemia and hyperammonemia) |
| Endocrine | Transient diabetes insipidus (sometimes seen) |
Severe Complications
- DIC with vaginal bleeding or post-cesarean hemorrhage
- Acute pancreatitis (16% in one cohort)
- Ascites and pleural effusion
- Respiratory failure
- Acute renal failure (75% had creatinine ≥1.5 mg/dL in one series; 2% required dialysis)
- Sepsis/infection
- Rare: myocardial infarction, pulmonary fat emboli
Outcomes Data (cohort of 51 women)
-
Blood product transfusion: 55%
-
Pancreatitis: 16%
-
ICU admission: 14%
-
Maternal death: 4%
-
Stillbirth: 12%
-
Acute renal failure requiring average 8-9 days to return to baseline creatinine
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 1375
-
Sleisenger and Fordtran's GI and Liver Disease, p. 661
Diagnosis
Clinical Diagnosis: The Swansea Criteria
Diagnosis requires 6 or more of the following criteria in the absence of another explanation (validated in the UK national cohort):
Clinical:
| Criterion | Threshold |
|---|---|
| Vomiting | Present |
| Abdominal pain | Present |
| Polydipsia/polyuria | Present |
| Encephalopathy | Present |
Laboratory:
| Criterion | Threshold |
|---|---|
| Bilirubin | >0.8 mg/dL (>14 µmol/L) |
| Hypoglycemia | <70 mg/dL (<4 mmol/L) |
| Uric acid | >5.7 mg/dL (>340 µmol/L) |
| Leukocytosis | >11 × 10⁹/L |
| ALT or AST | >42 U/L |
| Ammonia | >47 µmol/L |
| Coagulopathy | PT >14 s OR APTT >34 s |
Other:
| Criterion | Threshold |
|---|---|
| Ascites or bright liver on ultrasound | Present |
| Microvesicular steatosis on liver biopsy | Present |
(Modified from Knight et al., Gut 2008)
Typical Lab Profile
- AST/ALT: Moderately elevated (typically ~750 U/L) - rarely very high or even normal
- Bilirubin: Elevated (jaundice in majority)
- PT/aPTT: Prolonged
- Fibrinogen: Decreased (hypofibrinogenemia)
- Platelet count: May be low (thrombocytopenia)
- Creatinine/BUN: Elevated (renal dysfunction)
- Uric acid: Elevated
- Glucose: Low (hypoglycemia - critical finding)
- Ammonia: Elevated
- WBC: Leukocytosis
Imaging
- Hepatic ultrasound may show increased echogenicity ("bright liver") consistent with steatosis
- CT scan can confirm hepatic steatosis
- Imaging is important to exclude hepatic hematoma, rupture, and infarction
Liver Biopsy
-
Usually unnecessary if clinical and laboratory features are typical
-
Indicated when: the obstetrician has reservations about delivery, or diagnosis remains uncertain
-
Coagulopathy may necessitate transvascular (transjugular) sampling rather than percutaneous
-
Histologic results are pathognomonic when present
-
Comprehensive Clinical Nephrology 7th Ed., p. 633-634
-
Yamada's Textbook of Gastroenterology, p. 831
Differential Diagnosis
AFLP exists on a spectrum with other severe pregnancy-related liver diseases and must be distinguished from:
| Condition | Key Distinguishing Features |
|---|---|
| HELLP Syndrome | Hemolysis (elevated LDH, low haptoglobin, schistocytes), thrombocytopenia <100 × 10⁹/L; coagulation studies usually normal; occurs with preeclampsia |
| Severe Preeclampsia | Hypertension, proteinuria; transaminases elevated but usually not as severely; no hypoglycemia |
| Viral Hepatitis (Hep E, HSV) | Serologic testing positive; Hep E may be severe in pregnancy; HSV may be anicteric with herpetic lesions |
| Toxic/Drug-Induced | History of valproate, IV tetracycline; similar microvesicular pattern |
| Intrahepatic Cholestasis of Pregnancy | Pruritus dominant, bile salts elevated, transaminases mildly elevated, no liver failure |
| Hyperemesis Gravidarum | Earlier in pregnancy (1st trimester), no jaundice, no coagulopathy |
| Budd-Chiari Syndrome | Hepatic vein thrombosis on Doppler imaging |
Importantly, AFLP, HELLP, and severe preeclampsia all mandate prompt delivery, so the practical urgency to distinguish them is somewhat reduced - though recognizing hepatic hematoma and rupture is critically important.
The co-occurrence of preeclampsia and DIC in AFLP patients means they may simultaneously meet HELLP criteria, blurring the distinction.
Management
Principles
AFLP is a true obstetric emergency. Once diagnosed, management priorities are:
- Expedited delivery - the definitive treatment
- Aggressive supportive care in an ICU or high-dependency unit
- Correction of coagulopathy
- Prevention and treatment of complications
Step-by-Step Management
Immediate stabilization:
- Admit to ICU/high-dependency care
- Multidisciplinary team: maternal-fetal medicine obstetricians, intensivists, hepatologists, neonatologists, anesthesiologists
- Establish IV access; continuous monitoring of maternal vital signs and fetal status
Delivery:
- Prompt delivery is imperative regardless of gestational age
- Mode: Cesarean section is common (74% in the UK cohort), though vaginal delivery is not contraindicated if clinically feasible
- Coagulopathy must be corrected prior to delivery to reduce hemorrhagic risk
Coagulopathy management:
- Fresh frozen plasma (FFP) - to replace clotting factors
- Cryoprecipitate - for fibrinogen replacement
- Platelet transfusions as needed
- Blood products as required (55% of patients required transfusion)
Metabolic correction:
- Hypoglycemia: Continuous dextrose infusion; frequent glucose monitoring (at-risk patients with altered CNS function should be presumed hypoglycemic until proven otherwise)
- Hyperammonemia: Treat encephalopathy
Renal support:
- Monitor urine output and creatinine closely
- Renal replacement therapy (dialysis) in severe AKI (~2% require)
- Pathology multifactorial: hepatorenal syndrome, TMA, preeclampsia co-existing
Respiratory support:
- Mechanical ventilation as needed for respiratory failure
Other:
- Treat/prevent infection (sepsis is a major complication)
- Monitor for pancreatitis
Postpartum Course
-
Disease typically remits after delivery with no residual hepatic or renal impairment
-
Recovery of renal function takes average 8-9 days
-
Patients may remain critically ill for days-weeks postpartum while hepatic function recovers
-
Liver transplantation has been required in rare refractory cases (1 case in the UK cohort)
-
Sleisenger and Fordtran's GI and Liver Disease, p. 662
-
Creasy & Resnik's Maternal-Fetal Medicine, p. 1375
Fetal and Neonatal Implications
- Perinatal mortality: Historically high (7-58% in early retrospective series); in the UK prospective cohort: 104 per 1,000 births (approximately 10%)
- Stillbirth risk: ~12%
- Neonatal LCHAD screening: Infants born after AFLP-affected pregnancies should be screened for fatty acid oxidation disorders including LCHAD deficiency - deficient babies develop nonketotic hypoglycemia and obtundation after birth
- Genetic testing: In all cases of AFLP, the mother, father, and child should be tested for the G1528C LCHAD mutation
- Prenatal genetic diagnosis is feasible via chorionic villus sampling in families with known mutations
Recurrence and Future Pregnancies
- Recurrence of AFLP in subsequent pregnancies has been documented, particularly in women with confirmed LCHAD deficiency
- Women with AFLP in one pregnancy are not universally at risk for recurrence, but counseling and close monitoring in future pregnancies is warranted
- Some cases have resolved normally in subsequent pregnancies even after prior AFLP
Prognosis
| Era | Maternal Mortality | Perinatal Mortality |
|---|---|---|
| Historical (early series) | 12-18% | 7-58% |
| Modern (UK national cohort) | ~2% (1/57 cases died) | ~10% |
| With prompt diagnosis + delivery | Near 100% survival | <7% |
The dramatic improvement in outcomes is attributed to:
- Earlier recognition (Swansea criteria)
- Availability of intensive care and liver support
- Multidisciplinary obstetric management
- Blood product availability
Key Points Summary
| Feature | Detail |
|---|---|
| Incidence | ~1/6,700 to 1/16,000 pregnancies |
| Timing | 34-37 weeks gestation (third trimester) |
| Core mechanism | Fetal LCHAD deficiency → toxic fatty acid metabolites → maternal hepatotoxicity |
| Histology | Microvesicular steatosis, zone 3 predominant, no significant necrosis |
| Diagnosis | Swansea criteria (≥6/11 criteria) |
| Key lab | Hypoglycemia, coagulopathy, elevated transaminases, renal dysfunction |
| Treatment | Immediate delivery + ICU supportive care |
| Prognosis | Excellent with early diagnosis; near 100% maternal survival |
| Recurrence | Possible, especially with LCHAD deficiency |
| Neonatal | Screen infant for LCHAD; G1528C mutation testing of family |
Recent Evidence Update
A 2022 review in Gastroenterology by Terrault & Williamson emphasizes that AFLP's importance lies in significant maternal and fetal morbidity/mortality, that expeditious diagnosis is critical, and that a multidisciplinary approach (midwives, MFM specialists, anesthetists, neonatologists, hepatologists) is essential - consistent with textbook guidance. No major therapeutic changes since the foundational management principles were established (PMID: 35276220).
Sources:
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease, pp. 660-662
- Creasy & Resnik's Maternal-Fetal Medicine: Principles and Practice, p. 1375
- Comprehensive Clinical Nephrology, 7th Ed., pp. 633-634
- Yamada's Textbook of Gastroenterology, 7th Ed., pp. 830-831
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 1443-1445
- Goldman-Cecil Medicine (AFLP entry)
- Terrault NA, Williamson C. Pregnancy-Associated Liver Diseases. Gastroenterology 2022;162(7):1947-1963. [PMID: 35276220]
This is a shared conversation. Sign in to Orris to start your own chat.