Neuromyelitis optica spectrum disorders
Reading File
Finding Sources
Finding Sources
Searching PubMed
"neuromyelitis optica spectrum disorder" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material from multiple authoritative textbooks. Let me compile the complete, well-structured response.
Neuromyelitis Optica Spectrum Disorders (NMOSD)
Historical Background & Definition
NMOSD is an autoimmune inflammatory CNS disorder first described by Clifford Albutt (1870) and codified by Devic and Gault in the 1890s as a syndrome of optic neuritis (ON) combined with myelitis. Until 2004, it was widely regarded as a severe variant of MS. The discovery of the aquaporin-4 antibody (AQP4-IgG) by Lennon and colleagues at the Mayo Clinic definitively established NMOSD as a separate disease entity. - Adams and Victor's Principles of Neurology, 12th Ed., p. 940
The term NMOSD (Neuromyelitis Optica Spectrum Disorders) now encompasses the full range of clinical presentations linked to AQP4-IgG seropositivity, beyond just the classic optic nerve + spinal cord phenotype. - Bradley and Daroff's Neurology in Clinical Practice
Pathophysiology
Target antigen: AQP4 (Aquaporin-4), a transmembrane water channel protein expressed at astrocytic foot processes at the blood-brain barrier (BBB). - Bradley and Daroff's Neurology in Clinical Practice
Mechanism:
- AQP4-IgG (an IgG1 autoantibody) binds to AQP4 on astrocytic endfeet at capillaries, the pia, and Virchow-Robin spaces, and around the central canal of the spinal cord.
- This triggers complement activation (C5b-9 membrane attack complex), producing a "rim and rosette" pattern of immune complexes.
- The result is primary inflammatory astrocytopathy with secondary demyelination - distinct from MS which is primarily oligodendrocyte/myelin-targeted.
- Inflammatory infiltrate includes T cells, B cells, macrophages, and uniquely for NMOSD: neutrophils and eosinophils.
- Areas of damage show necrosis, vascular immunoglobulin and complement deposition, and loss of AQP4 expression.
This humoral immunopathology (vs. MS's predominantly cellular mechanism) explains the differential treatment response: MS DMTs like natalizumab and interferon-beta can paradoxically worsen NMOSD. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Epidemiology
| Feature | NMOSD | MS (comparison) |
|---|---|---|
| Prevalence | 0.5-4.4/100,000 | 50-100x more common |
| Female:Male ratio | 9:1 (AQP4+) | ~3:1 |
| Age of onset | ~40 years (median) | ~28-30 years |
| Ethnicity | Non-Caucasian predominance (Hispanic, African, Asian) | Caucasian predominance |
| Course | Relapsing in ~85% | Relapsing-remitting, then secondary progressive |
- Bradley and Daroff's Neurology in Clinical Practice; Grainger & Allison's Diagnostic Radiology
Core Clinical Characteristics (Wingerchuk 2015 Criteria)
There are 6 core clinical characteristics - at least one is required for diagnosis in AQP4-IgG seropositive patients:
-
Optic Neuritis (ON) - typically severe, often bilateral, involving long segments of the optic nerve and frequently the posterior optic pathway (chiasm, optic tracts). Recovery may be incomplete.
-
Acute Myelitis - characteristically a longitudinally extensive transverse myelitis (LETM): contiguous spinal cord lesion spanning ≥3 vertebral segments, often extending from cervical to thoracic cord. Affects motor, sensory, and bowel/bladder function.
-
Area Postrema Syndrome - intractable nausea, vomiting, or hiccups due to dorsal medulla involvement; presenting symptom in up to 10% of patients.
-
Acute Brainstem Syndrome - double vision, dysphagia, ataxia, oculomotor dysfunction, respiratory compromise.
-
Symptomatic Narcolepsy or Acute Diencephalic Syndrome - with typical NMOSD diencephalic MRI lesions (hypothalamic involvement causes anorexia, hypothermia, hypersomnia, inappropriate diuresis).
-
Symptomatic Cerebral Syndrome - with typical NMOSD brain lesions (corticospinal tract, periventricular).
- Grainger & Allison's Diagnostic Radiology (Table 58.4); Bradley and Daroff's Neurology in Clinical Practice (Table 80.5)
Diagnostic Criteria (Wingerchuk et al., Neurology, 2015)
AQP4-IgG Seropositive NMOSD
All three required:
- At least 1 core clinical characteristic
- Positive AQP4-IgG (cell-based assay strongly recommended; sensitivity 76.7%, specificity 99.8%)
- Exclusion of alternative diagnoses
AQP4-IgG Seronegative NMOSD (or unknown status)
All three required:
- At least 2 core clinical characteristics from ≥1 attack, with:
- At least one must be ON, LETM myelitis, or area postrema syndrome
- Dissemination in space (two or more different core characteristics)
- Fulfillment of additional MRI requirements
- Negative AQP4-IgG or unavailable
- Exclusion of alternative diagnoses
Investigations
CSF
- Pleocytosis >50 leukocytes/mm³ (often with neutrophils and eosinophils) is common
- Oligoclonal bands (OCBs): present in only 10-20% (vs. 80-90% in MS) - Bradley and Daroff's
AQP4-IgG
- Cell-based assay: sensitivity ~76-80%, specificity ~94-100%
MOG-IgG (anti-MOG)
- Found in a subset of AQP4-seronegative patients
- Also seen with ADEM, isolated ON, LETM
- Associated with younger age of onset, equal sex ratio, more often monophasic course, better prognosis
MRI Findings
Spinal Cord
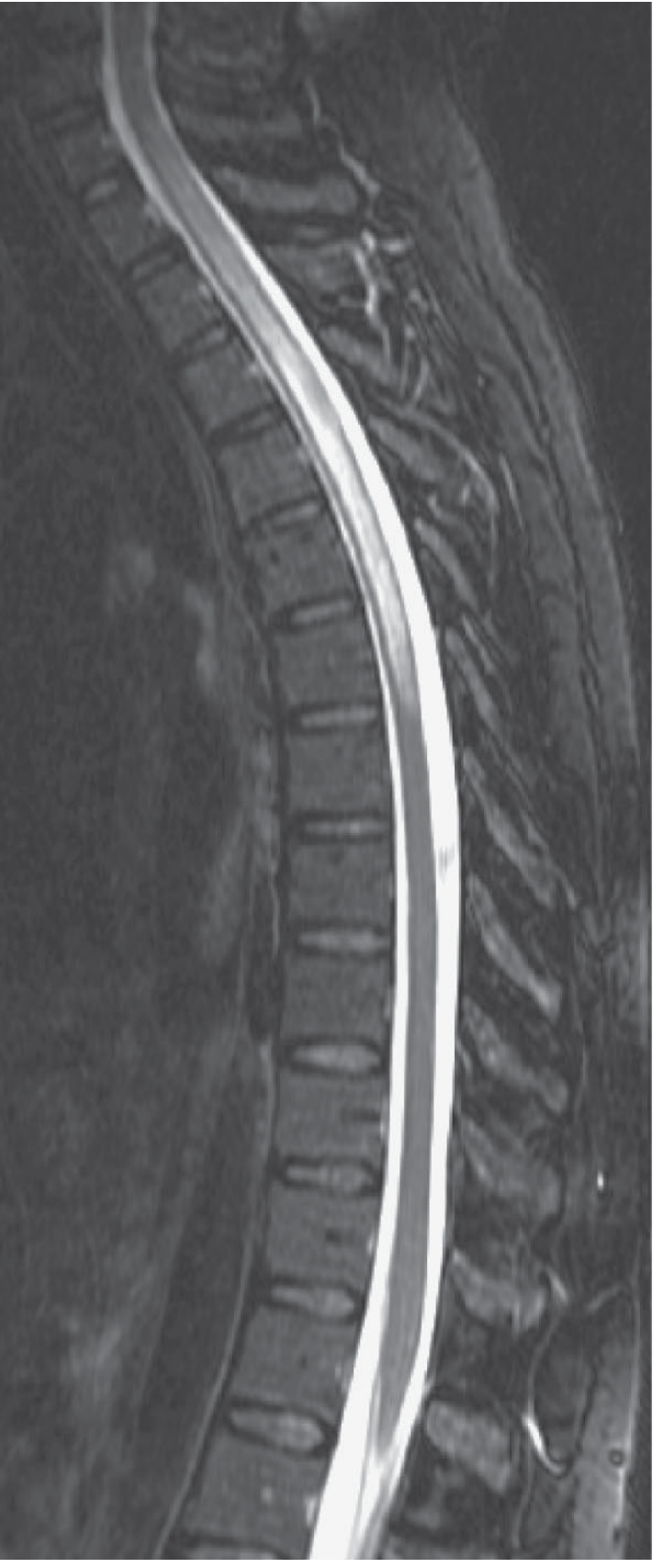
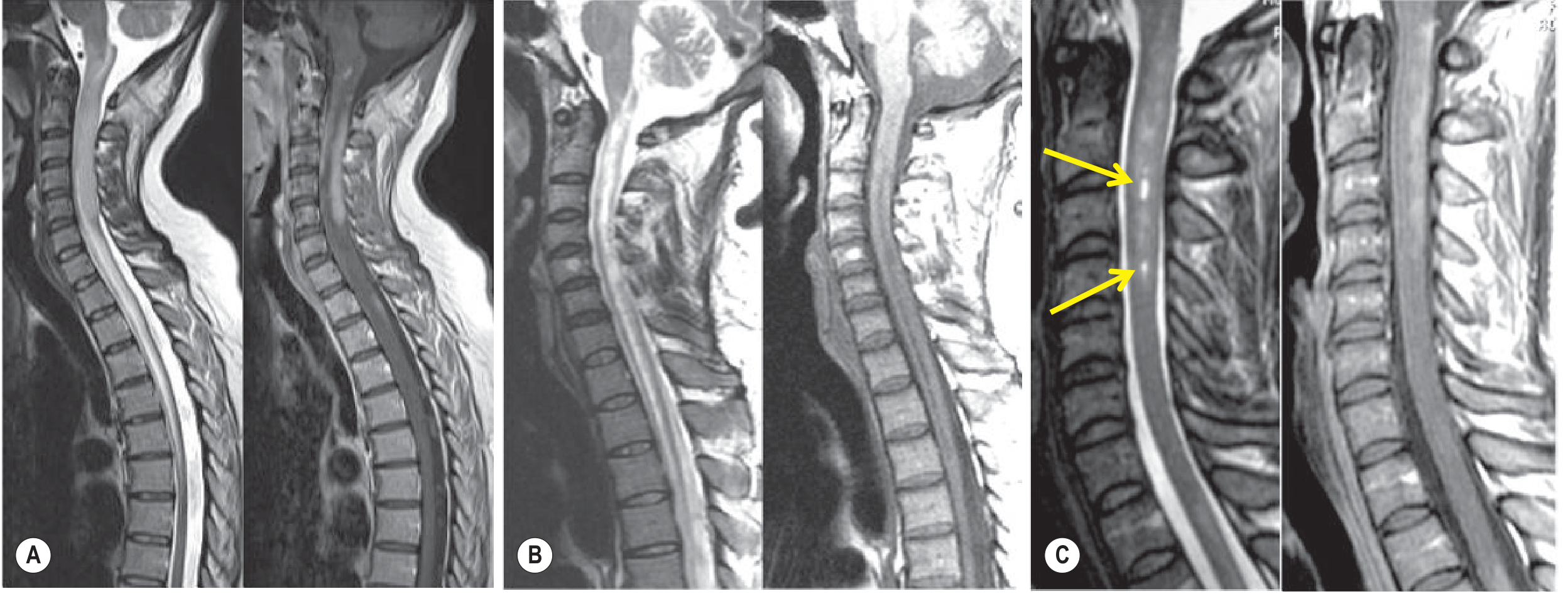
Key spinal cord MRI features:
- LETM (≥3 contiguous vertebral segments) - most specific finding
- Central location (gray matter predominance) on axial
- Bright spotty sign (T2 hyperintense foci) - differentiates from MS
- T1 hypointensity ("pseudo-syrinx") indicates necrosis
- Peripheral/rim contrast enhancement in acute phase
- Progression to atrophy and syrinx formation
Optic Nerve
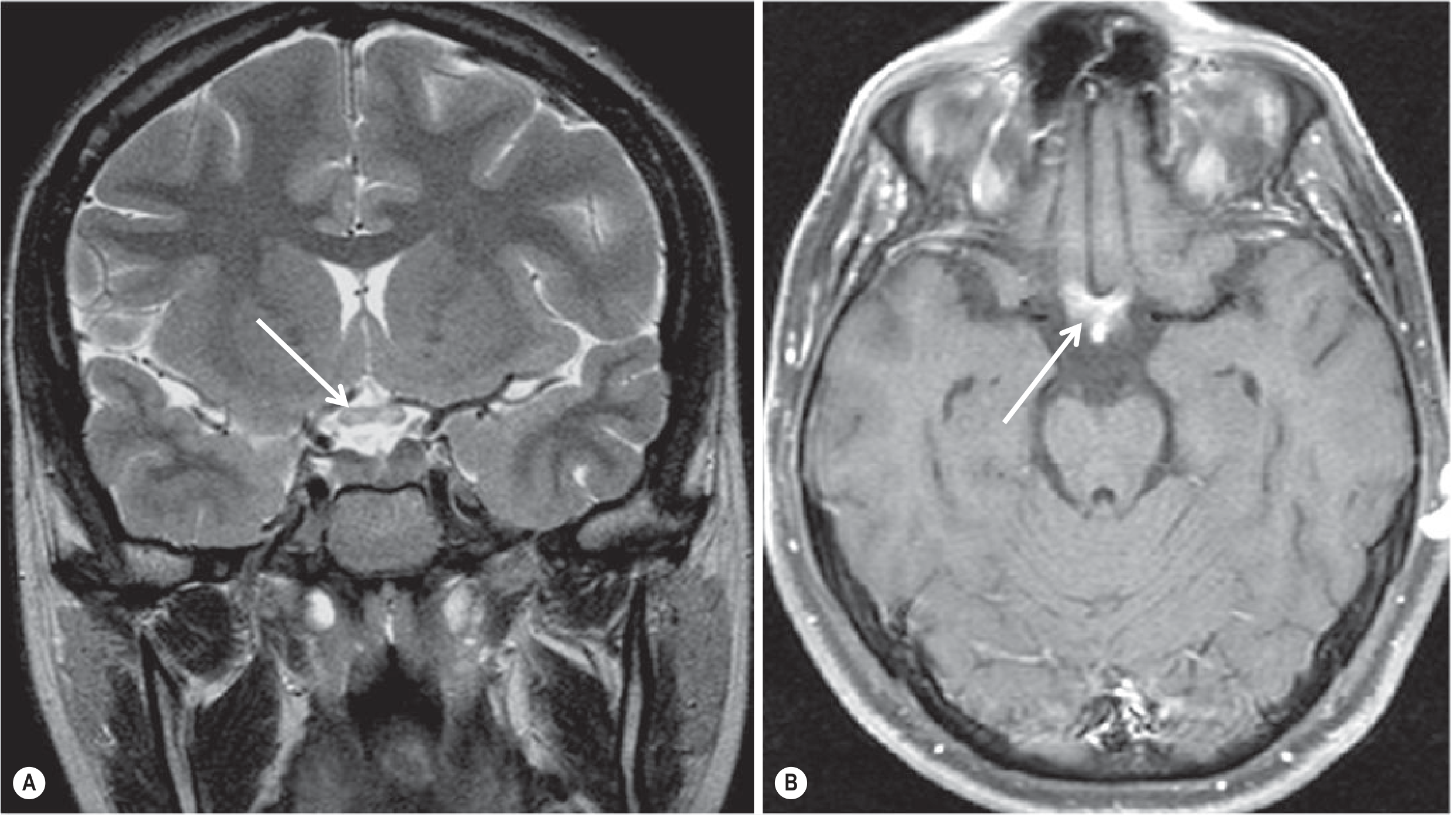
- Bilateral and longitudinally extensive, typically >1/2 optic nerve length
- Predominantly posterior optic pathway: intracranial optic nerves, chiasm, optic tracts
- Acute: T2 hyperintensity + gadolinium enhancement; chronic: atrophy
Brain
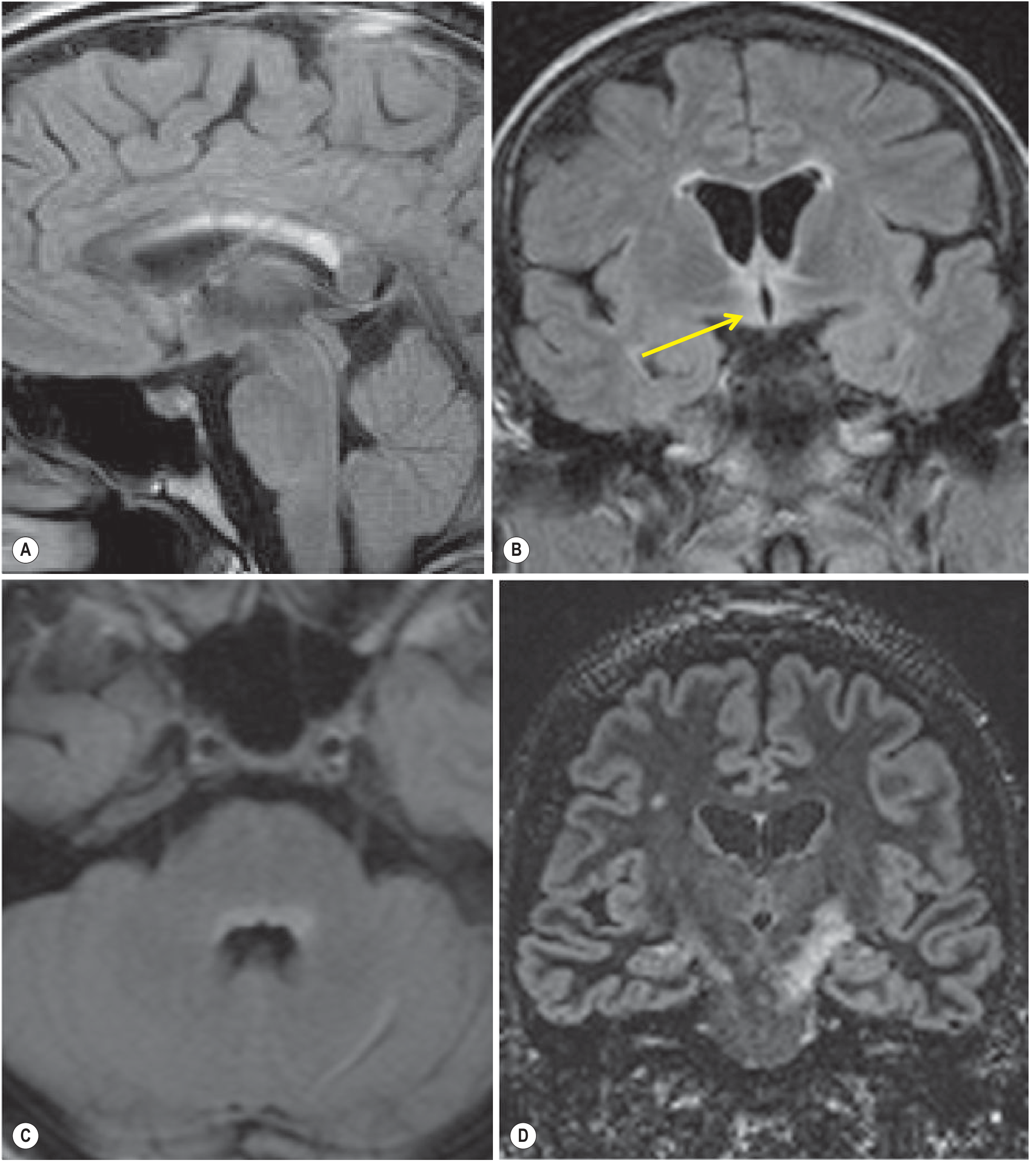
Brain MRI abnormalities parallel sites of high AQP4 expression (circumventricular organs):
- Hypothalamus, periependymal areas, corpus callosum (long "pencil-thin" lesions, unlike MS's short ovoid ones)
- Dorsal brainstem adjacent to 4th ventricle (area postrema)
- Periventricular/periaqueductal white matter
- Large hemispheric white matter lesions (atypical for MS)
NMOSD vs. MS: Key Differentiators
| Feature | NMOSD | MS |
|---|---|---|
| Target cell | Astrocyte (AQP4) | Oligodendrocyte |
| Immunopathology | Humoral (B cell/complement) | Cellular (T cell) |
| OCBs in CSF | 10-20% | 80-90% |
| Spinal cord lesions | LETM (≥3 segments), central, gray matter | Short (<2 segments), peripheral, white matter |
| Optic neuritis | Bilateral, severe, posterior, chiasmal | Unilateral, recovers well |
| Brain lesions | Periependymal, circumventricular, atypical | Periventricular, Dawson's fingers, juxtacortical |
| Sex ratio | 9:1 F:M | 3:1 F:M |
| MS DMTs | Contraindicated (may worsen) | Beneficial |
MOG Antibody-Associated Disease (MOGAD) - Key Distinctions
MOG-IgG positive NMOSD deserves separate mention:
- Demographics: More common in children/young adults; nearly equal sex ratio (vs. female predominance in AQP4+)
- Course: Often monophasic or episodic; persistent high titers predict relapsing course
- ON: Disc edema prominent; long optic nerve involvement; often bilateral
- Myelitis: Lower in spinal cord, can be short-segment or LETM; area postrema syndrome less common
- Prognosis: Better than AQP4+ NMOSD; less severe neurological deficits; but bowel/bladder/erectile dysfunction can be permanent
- Pathology: Demyelination targets oligodendrocytes (not astrocytes); no complement deposition
Clinical Course & Prognosis
- ~85% relapsing course with severe, stepwise disability accumulation
- Unlike MS, progressive course without relapses is rare
- Individual attacks tend to be more severe: complete blindness or paralysis from single attacks is common
- Respiratory failure from high cervical myelitis is a major cause of death
- AQP4-seropositive patients: more severe attacks, higher relapse rate, worse outcomes vs. seronegative
- Untreated: ~50% of patients become blind in one eye or need walking support within 5 years
Treatment
Acute Attack Management
- High-dose IV methylprednisolone - 1 g/day for 5-10 days, followed by oral prednisone taper
- Plasma Exchange (PLEX) - 5-7 exchanges at 1.5 plasma volumes - used for:
- Severe attacks not responding to steroids
- Attacks with serious neurological deficit (early use improves outcomes)
- May be used as first-line in severe attacks
- IVIG - alternative for incomplete steroid recovery
Relapse Prevention (Long-term Immunotherapy)
FDA-approved monoclonal antibodies (AQP4-seropositive NMOSD):
| Drug | Mechanism | Attack Risk Reduction | Route |
|---|---|---|---|
| Eculizumab (Soliris) | C5 complement inhibitor | 94% (add-on to IS) | IV q2 weeks |
| Ravulizumab (Ultomiris) | C5 complement inhibitor (long-acting) | 100% (50-week obs.) | IV q8 weeks |
| Inebilizumab (Upilzna) | Anti-CD19 B-cell depleter | 77-78% (monotherapy) | IV q6 months |
| Satralizumab (Enspryg) | Anti-IL-6 receptor | 74-78% (mono or add-on) | SC q4 weeks |
Harrison's Principles of Internal Medicine 22E (2025), Table 456-2
Preferred approach (Harrison's): Start with inebilizumab or satralizumab; complement inhibitors as second-line for non-responders.
Off-label agents (historically used, especially AQP4-seronegative):
- Rituximab (anti-CD20 B-cell depletion)
- Mycophenolate mofetil
- Azathioprine
- Mitoxantrone, cyclophosphamide (limited use)
Important warning: MS disease-modifying therapies (natalizumab, interferons, fingolimod) should NOT be used - they may worsen NMOSD attacks.
Monitoring for approved agents:
- Eculizumab/ravulizumab: Meningococcal vaccination mandatory; monitor for meningococcal infection (boxed warning)
- Inebilizumab: Monitor serum IgG and CBC (neutropenia risk)
- Satralizumab: Screen for HBV, TB; monitor LFTs and CBC; weight gain in ~30%
Recent Evidence
A 2025 Bayesian network meta-analysis (Immunotherapies in neuromyelitis optica, PMID 40775082, J Neurol 2025) further evaluated the comparative efficacy of immunotherapies in NMOSD. A 2025 meta-analysis (MRI lesion distribution in MS vs. NMOSD vs. MOGAD, PMID 40889881, J Neurol Neurosurg Psychiatry 2025) confirmed distinct MRI lesion distribution profiles that help differentiate these three conditions, supporting the 2015 diagnostic criteria framework.
Differential Diagnosis Summary
- Multiple sclerosis - OCBs, periventricular lesions, no AQP4-IgG, short cord lesions
- ADEM - typically monophasic, post-infectious, pediatric, MOG-IgG common
- Systemic autoimmune disease (SLE, Sjögren's) - may coexist or mimic; always test AQP4-IgG in these patients presenting with myelitis/ON
- Spinal cord infarction - DWI positive, vascular risk factors
- Paraneoplastic myelopathy - search for occult malignancy if atypical
- Sarcoidosis - leptomeningeal enhancement, systemic features
This is a shared conversation. Sign in to Orris to start your own chat.