A 65-year-old man is evaluated for a generalized, intensely pruritic eruption that has been slowly progressing over the last 6 months. He has been treated with topical corticosteroids for 4 months for widespread eczema without relief of pruritus or change in clinical appearance. He has never had a skin biopsy. He does not have a personal or family history of asthma, atopic dermatitis, allergic rhinitis, or psoriasis. On physical examination, temperature is 37.5 °C, blood pressure is 135/85 mm Hg, pulse rate is 84/min, and respiration rate is 14/min. Skin examination reveals erythema with scale affecting greater than 90% of the body surface area. Alopecia, nail dystrophy, and ectropion are present. There is thickening and fissuring of the skin on the palms and soles. Bilateral axillary and inguinal lymphadenopathy is present. The mucous membranes are not involved. QUESTIONS 1. What is the most likely diagnosis? 2. List 3 differential diagnoses 3. What is the pathophysiology? 4. What are the predisposing factors? 5. How do you confirm the diagnosis? 6. What is the treatment?
Clinical Case Analysis: Erythroderma (Exfoliative Dermatitis)
1. Most Likely Diagnosis
- Age and sex: 65-year-old male - fits the classic demographic (peak onset 6th-7th decade, male predominance 2:1 to 4:1)
- Intense, intractable pruritus: A hallmark of Sézary syndrome
- Absence of atopic history: No personal or family history of atopy (asthma, atopic dermatitis, allergic rhinitis) - making atopic dermatitis-driven erythroderma unlikely
- No psoriasis history: No personal or family history of psoriasis
- Failure of topical corticosteroids: 4 months of treatment with zero response strongly suggests an underlying neoplastic process
- Bilateral axillary and inguinal lymphadenopathy: Lymph node involvement indicates systemic disease
- Chronic progression (6 months): Gradual onset is characteristic of CTCL, not acute drug reactions
- Alopecia, nail dystrophy, ectropion, and palmoplantar keratoderma: Classic constellation of Sézary syndrome features
2. Three Differential Diagnoses
| Diagnosis | Key Distinguishing Features |
|---|---|
| 1. Erythrodermic Psoriasis | Personal/family history of psoriasis; responds to corticosteroid withdrawal; silvery scaling; nail pitting; no lymphadenopathy; Auspitz sign |
| 2. Drug-induced Erythroderma | More acute onset; identifiable culprit drug; usually resolves 2-6 weeks after withdrawal; may include eosinophilia (DRESS); no bilateral lymphadenopathy |
| 3. Atopic Dermatitis-related Erythroderma | Personal/family atopic history; elevated IgE; eosinophilia; onset often younger; responds to corticosteroids and emollients |
3. Pathophysiology
- The number of germinative keratinocytes and their mitotic rate is markedly increased in erythrodermic skin
- The transit time of cells through the epidermis is shortened
- Scales consist of material normally retained by the skin (nucleic acids, amino acids, soluble proteins)
- Daily scale loss increases from a normal 500-1000 mg to 20-30 g/day
- In acute erythroderma, desquamated material has marginal metabolic significance
- In chronic erythroderma, protein loss becomes significant - leading to hypoalbuminemia and anemia of chronic disease
- Massive cutaneous vasodilation disrupts normal heat regulation, causing hypothermia or hyperthermia
- Transepidermal water loss is greatly increased, leading to dehydration and electrolyte imbalances
- Malignant T-cells (Sézary cells) are CD4+ memory T-cells with cerebriform nuclei
- These cells traffic between the skin, lymph nodes, and peripheral blood
- They produce a predominantly Th2 cytokine profile (IL-4, IL-5, IL-13), explaining the intense pruritus
- Loss of normal pan-T-cell markers (CD7, CD26) and an elevated CD4:CD8 ratio (>10:1) are characteristic
- Hypothermia (vasodilation + evaporative heat loss)
- Peripheral edema and cardiac failure (high-output state from cutaneous vasodilation)
- Hypoalbuminemia and cachexia (protein loss via scale)
- Secondary infections (Staphylococcus aureus, streptococcal)
- Capillary leak syndrome, acute respiratory distress syndrome, and sepsis
4. Predisposing Factors
- Pre-existing dermatoses (psoriasis, atopic dermatitis, seborrheic dermatitis, contact dermatitis)
- Drug exposure (allopurinol, anticonvulsants, sulfonamides, penicillins, gold, cimetidine, isoniazid, omeprazole - over 130 drugs implicated)
- Underlying malignancy (CTCL, Hodgkin's lymphoma, solid organ cancers)
- HIV infection (especially drug-induced forms)
- Age-related immune senescence (peak incidence 6th-7th decade)
- Male sex (striking male predominance)
- Older age (median age ~60-65 years)
- Immune dysregulation (chronic antigen stimulation, T-cell clonal expansion)
- No clear environmental or genetic trigger has been definitively identified; however, molecular studies show consistent TCR gene rearrangements suggesting a clonal origin
- The striking male predominance in idiopathic erythroderma (which often harbors occult CTCL) suggests possible hormonal or immune factors
5. How to Confirm the Diagnosis
- Multiple biopsies from different anatomical sites
- Look for: cerebriform pleomorphic lymphocytes, band-like and lichenoid infiltrate, epidermal clustering of atypical cells (Pautrier microabscesses)
- Note: Cutaneous histology is non-diagnostic in at least 30% of Sézary syndrome patients - repeat biopsies may be necessary
- Absolute Sézary cell count ≥1000 cells/mm³ in peripheral blood
- CD4:CD8 ratio ≥10:1 by flow cytometry (caused by expansion of circulating malignant CD4+ T cells or loss of pan-T-cell markers)
- Evidence of a T-cell clone by Southern blot or PCR (T-cell receptor gene rearrangement)
- Chromosomally abnormal T-cell clone
- CD4+/CD7- or CD4+/CD26- phenotype of circulating T cells
- CD4:CD8 ratio
- Loss of pan-T-cell markers
- Bilateral axillary and inguinal lymph node biopsy/FNA
- CT scan of chest/abdomen/pelvis to stage disease
- CBC with differential (lymphocytosis, eosinophilia)
- Serum LDH (elevated in high tumor burden)
- Serum IgE (elevated in atopic erythroderma)
- Serum albumin, electrolytes (systemic complications)
- Patch testing if drug reaction suspected
- HTLV-1 serology (exclude adult T-cell leukemia/lymphoma)
6. Treatment
- Emollients and wet dressings to restore barrier function and reduce TEWL
- Tepid baths to prevent hypothermia
- Nutritional support (high-protein diet, albumin replacement if needed)
- Antihistamines for pruritus (often inadequate alone in CTCL)
- Antibiotics for secondary skin infections (S. aureus)
- Temperature-controlled environment
- Topical corticosteroids (limited role in Sézary syndrome - as shown by this case, they failed)
- Topical nitrogen mustard (mechlorethamine) - applied to affected skin
- PUVA (psoralen + UVA) or narrowband UVB phototherapy
- Total skin electron beam therapy (TSEBT) - highly effective for diffuse skin disease
- Extracorporeal photopheresis (ECP): First-line for Sézary syndrome - involves collecting peripheral blood, exposing mononuclear cells to 8-methoxypsoralen + UVA, then reinfusing; immunomodulatory mechanism
- Retinoids: Bexarotene (RXR-selective retinoid) - FDA-approved for CTCL; often used with ECP
- Interferon-alpha (IFN-α): Immunomodulatory, often combined with ECP or PUVA
- Histone deacetylase inhibitors: Vorinostat, romidepsin - FDA-approved for relapsed/refractory CTCL
- Mogamulizumab: Anti-CCR4 monoclonal antibody - approved specifically for CTCL including Sézary syndrome; targets malignant T-cells expressing CCR4
- Low-dose methotrexate: Modest activity, used in resource-limited settings
- Alemtuzumab (anti-CD52): Effective but significant immunosuppression risk
- The only potentially curative option in eligible patients with advanced/refractory disease
- Treat high-output cardiac failure, sepsis, and electrolyte imbalances as they arise
- Avoid systemic corticosteroids in CTCL-suspected cases (may mask diagnosis and worsen disease long-term)
- This case is a classic presentation of Sézary syndrome - the leukemic variant of CTCL
- The combination of erythroderma + lymphadenopathy + alopecia + nail dystrophy + ectropion + palmoplantar keratoderma + treatment-resistant intense pruritus in an elderly male is the textbook picture
- Absence of atopic/psoriatic history and failure of topical steroids for 4 months are strong red flags against benign causes
- Skin biopsy has never been performed in this patient - this is the immediate next step
- Prognosis is poor (similar to nodal-stage CTCL); median survival for Sézary syndrome is approximately 2-4 years
A 65-year-old man is evaluated for a generalized, intensely pruritic eruption that has been slowly progressing over the last 6 months. He has been treated with topical corticosteroids for 4 months for widespread eczema without relief of pruritus or change in clinical appearance. He has never had a skin biopsy. He does not have a personal or family history of asthma, atopic dermatitis, allergic rhinitis, or psoriasis. On physical examination, temperature is 37.5 °C, blood pressure is 135/85 mm Hg, pulse rate is 84/min, and respiration rate is 14/min. Skin examination reveals erythema with scale affecting greater than 90% of the body surface area. Alopecia, nail dystrophy, and ectropion are present. There is thickening and fissuring of the skin on the palms and soles. Bilateral axillary and inguinal lymphadenopathy is present. The mucous membranes are not involved. QUESTIONS 1. What is the most likely diagnosis? 2. List 3 differential diagnoses 3. What is the pathophysiology? 4. What are the predisposing factors? 5. How do you confirm the diagnosis? 6. What is the treatment?
Clinical Case: Sézary Syndrome
1. Most Likely Diagnosis
| Feature in this patient | Significance |
|---|---|
| 65-year-old male | Peak demographic: male:female ratio 2:1 to 4:1, peak onset 6th-7th decade |
| Intensely pruritic, >90% BSA erythema + scale | Core definition of erythroderma |
| Alopecia, nail dystrophy, ectropion, palmoplantar keratoderma | Classic constellation of Sézary syndrome |
| Bilateral axillary AND inguinal lymphadenopathy | Systemic lymph node involvement - most common extracutaneous finding (~50% of erythroderma patients), but bilateral axillary + inguinal together strongly suggests neoplastic cause |
| No atopic history (no asthma, no AD, no allergic rhinitis) | Rules out atopic dermatitis as the driver |
| No personal/family psoriasis history | Rules out psoriatic erythroderma |
| 4 months of topical corticosteroids - zero response | Benign erythroderma typically responds; CTCL does not - topical steroids may even worsen CTCL |
| Slowly progressive over 6 months | Indolent neoplastic course, not acute drug reaction |
| Never had a skin biopsy | Diagnosis yet to be confirmed |
2. Three Differential Diagnoses
- Most common cause of erythroderma overall (20% of cases)
- Typically preceded by known psoriatic plaques; personal or family history of psoriasis usually present
- Nail changes (pitting, oil-drop sign, onycholysis) rather than dystrophy; no ectropion
- Trigger often identifiable: corticosteroid withdrawal, systemic infection, biologics
- After generalization, typical psoriatic features may be lost - making distinction difficult without biopsy
- This patient has no personal or family psoriasis history - making this less likely
- 19% of erythroderma cases; accounts for over 130 causative drugs (allopurinol, anticonvulsants, sulfonamides, penicillins, gold, NSAIDs, omeprazole)
- More acute onset; usually resolves 2-6 weeks after drug withdrawal
- Associated features: fever, eosinophilia, lymphadenopathy (reactive rather than neoplastic)
- No drug history is identified in this case, and the 6-month chronic progressive course argues against it
- HIV-positive patients have a higher rate of drug-induced erythroderma
- 9% of erythroderma cases (most common within the dermatitis subgroup)
- Requires pre-existing or personal/family history of atopy (asthma, allergic rhinitis, AD)
- Elevated serum IgE and peripheral eosinophilia are characteristic
- Pruritus is intense; secondary excoriations and prurigo-like lesions common
- This patient has explicitly no atopic history - making this diagnosis unlikely
Other causes worth noting: pityriasis rubra pilaris (PRP - characterized by "islands of sparing," orange nutmeg-grater follicular papules on dorsal fingers), pemphigus foliaceus, Hodgkin lymphoma (fever, splenomegaly, elevated ESR), and idiopathic erythroderma (~25% of cases, some of which eventually declare as CTCL).
3. Pathophysiology
A. Epidermal Kinetics (common to all erythroderma)
- In acute erythroderma, scale loss has marginal metabolic significance
- In chronic erythroderma (as in this case), protein loss becomes clinically significant, causing hypoalbuminemia and contributing to anemia of chronic disease
B. Vascular and Systemic Consequences
- Massive cutaneous vasodilation increases blood flow to the skin, raising total cardiac output - risk of high-output cardiac failure (particularly dangerous in the elderly)
- Pedal/pretibial edema occurs in ~50% of patients from hypoalbuminemia and extracellular fluid shift
- Thermoregulatory disruption: excessive heat loss leads to compensatory hypermetabolism, rigor-like chills, and eventually cachexia
- Greatly increased transepidermal water loss leads to dehydration and electrolyte imbalance
- Secondary infection by Staphylococcus aureus or streptococci is common; can progress to sepsis
C. Sézary Syndrome-Specific Pathogenesis (Robbins Pathologic Basis of Disease)
- Sézary cells are CD4+ helper T-cells that home to skin via adhesion molecules - cutaneous lymphocyte antigen (CLA) and chemokine receptor CCR4
- These malignant T-cells have characteristic cerebriform nuclei (markedly infolded nuclear membrane) - visible in skin, peripheral blood, and lymph nodes
- They adopt a Th2 cytokine profile (IL-4, IL-5, IL-13), which explains:
- The intense, unrelenting pruritus
- Elevated serum IgE
- Peripheral eosinophilia
- Reduced delayed-type hypersensitivity (cutaneous anergy)
- Resistance to Fas-ligand and TNF-related apoptosis has been demonstrated, allowing clonal T-cell survival and expansion
- Unlike mycosis fungoides (where skin lesions progress through patch → plaque → tumor), in Sézary syndrome the skin lesions rarely proceed to tumefaction - the malignant cells traffic between skin, blood, and lymph nodes from the outset
4. Predisposing Factors
- Pre-existing dermatoses (psoriasis, atopic dermatitis, seborrheic dermatitis, contact dermatitis) - account for ~44% of cases
- Drug exposure (>130 drugs implicated)
- Underlying malignancy (CTCL, Hodgkin lymphoma, solid organ cancers)
- HIV infection (particularly drug-induced forms)
- Age-related immune senescence - peak incidence in the 6th to 7th decade
- Male sex (male:female ratio 2:1 to 4:1; even more striking in idiopathic/CTCL erythroderma)
- Older age (median ~60-65 years)
- Male predominance (striking; reason unclear but may involve hormonal or immune factors)
- Chronic antigenic stimulation leading to clonal T-cell expansion
- No definitive environmental or hereditary trigger has been identified
- Chromosomal aberrations are common in tumor cells (differ from the typical pattern of classic mycosis fungoides)
- Age-related immune senescence may impair normal T-cell surveillance, permitting clonal escape
5. How to Confirm the Diagnosis
A. Skin Biopsy (first and most important step)
- Multiple biopsies from different anatomical sites and on separate occasions
- Look for: cerebriform pleomorphic lymphocytes, band-like or lichenoid dermal infiltrate, clustering of atypical lymphocytes within the epidermis (Pautrier microabscesses)
- Histology pattern: spongiotic or lichenoid; may be entirely nonspecific - repeat biopsies are often necessary
B. Peripheral Blood Studies - ISCL/EORTC Diagnostic Criteria for Sézary Syndrome
- Absolute Sézary cell count ≥1,000 cells/mm³ in peripheral blood
- CD4:CD8 ratio ≥10:1 by flow cytometry (from expansion of malignant CD4+ T cells or loss of pan-T-cell markers)
- Evidence of a T-cell clone by Southern blot or PCR (T-cell receptor gene rearrangement)
- A chromosomally abnormal T-cell clone
C. Flow Cytometry (peripheral blood and skin)
- CD4+/CD7- or CD4+/CD26- phenotype of circulating T cells - highly characteristic
- Increased CD3+/CD4+ with loss of normal pan-T-cell markers
D. Lymph Node Evaluation
- Core or excisional biopsy of the most enlarged node (preferred over fine needle aspirate - need tissue architecture)
- CT or PET-CT scan first to identify the most FDG-avid/morphologically abnormal node
- Distinguish lymphomatous infiltration from reactive dermatopathic lymphadenopathy
E. Additional Investigations
| Test | Purpose |
|---|---|
| CBC with differential | Lymphocytosis (leukocytosis up to 30,000/mm³), eosinophilia |
| Serum LDH | Elevated with high tumor burden |
| Serum albumin, electrolytes | Assess systemic complications |
| Serum IgE | Elevated in atopic erythroderma; also elevated in Sézary due to Th2 skew |
| HTLV-1 serology | Exclude adult T-cell leukemia/lymphoma (clinically similar) |
| Chest/abdominal/pelvic CT | Full staging |
| Patch testing | If drug reaction remains on differential |
| Skin scrapings for mites | Exclude crusted scabies |
6. Treatment
A. General Supportive Care (for all erythroderma)
- Emollients and wet dressings to restore barrier function and reduce transepidermal water loss
- Tepid-to-warm baths and a temperature-controlled environment to combat thermoregulatory disruption
- Nutritional support: high-protein diet; albumin infusion if hypoalbuminemia is severe
- Antihistamines for pruritus (often inadequate alone in CTCL)
- Antibiotics for secondary S. aureus and streptococcal skin infections
- Avoid systemic corticosteroids in suspected CTCL - they may mask the diagnosis and lead to rebound worsening
B. Skin-Directed Therapies
- Topical nitrogen mustard (mechlorethamine) - applied to affected skin; limited utility in erythrodermic disease
- PUVA (psoralen + UVA) or narrowband UVB phototherapy
- Total skin electron beam therapy (TSEBT) - effective for diffuse skin disease; produces complete cutaneous responses and reduces peripheral blood tumor burden
C. Systemic Therapies for Sézary Syndrome
| Agent | Mechanism / Notes |
|---|---|
| Extracorporeal photopheresis (ECP) | First-line for Sézary syndrome. Peripheral blood mononuclear cells exposed ex vivo to 8-methoxypsoralen + UVA, then reinfused. Immunomodulatory; median survival 39-60 months. Often combined with retinoids or IFN-α |
| Bexarotene (retinoid) | RXR-selective retinoid, FDA-approved for CTCL. Often combined with ECP |
| Interferon-alpha (IFN-α) | Immunomodulatory; combined with ECP or PUVA |
| Low-dose methotrexate | ~50% response rate; overall survival ~101 months in responders (Andrews'); used when ECP unavailable |
| Mogamulizumab | Anti-CCR4 monoclonal antibody. Targets CCR4 expressed on malignant Sézary cells. FDA-approved for relapsed/refractory CTCL including Sézary |
| Vorinostat / Romidepsin | Histone deacetylase (HDAC) inhibitors; FDA-approved for relapsed/refractory CTCL |
| Alemtuzumab (anti-CD52) | Effective but profound immunosuppression; reserved for refractory cases |
D. Potentially Curative
- Allogeneic stem cell transplantation - the only potentially curative option in eligible patients with advanced/refractory Sézary syndrome
Prognosis
- Erythroderma is a clinical syndrome (not a diagnosis) - always search for the underlying cause
- Sézary syndrome = erythroderma + intense pruritus + lymphadenopathy + alopecia + nail dystrophy + ectropion + palmoplantar keratoderma in an elderly male = classic picture
- The absence of atopic/psoriasis history and failure of topical steroids for 4 months are the critical red flags pointing away from benign disease
- Skin biopsy has never been performed - this is the single most urgent next step
- Diagnosis requires peripheral blood flow cytometry + T-cell gene rearrangement studies in addition to biopsy, since histology is non-diagnostic in 30% of cases
- Never use systemic corticosteroids in suspected CTCL
SCENARIO 3 A 37-year-old female presents to the clinic complaining of amenorrhea. She has had normal menses until 8 months ago, when they became infrequent, and then stopped. She insists she cannot be pregnant, because she denies sexual activity “in years.” She believes she is going through “the change” but wants to know why she is reaching menopause at a much earlier age than other women she knows. On review of systems, she complains of headaches “for years” and recent onset of weakness and fatigue. She also complains of arthritis in the hip and knees, something she attributes to “getting old.” She denies other complaints. Physical exam results show that her vitals are normal. The patient is an adult female of average height, with a noticeably large jaw and hands. She is not wearing rings on her fingers (she reports that now that she is fat, they don’t fit any more). Her hair is thick and coarse, and hirsutism is present. Her thyroid gland is slightly enlarged, but regular in shape. No bruit or tenderness is present. The point of maximal impulse is displaced laterally, but the heart is regular with no murmurs. The rest of the exam is normal. QUESTIONS 1. What is the diagnosis and what features suggest the diagnosis in this patient? What other signs would one look for on the face and mouth. List 3. 2. 3. List any 5 of the complications. What is the next step in this patient? What other tests would you do and what would you see? List 3 4. 5. 6. What is the best treatment in this patient? Discuss other treatment options available. List any other 2 conditions which may present with increased growth hormone levels.
Clinical Case: Acromegaly
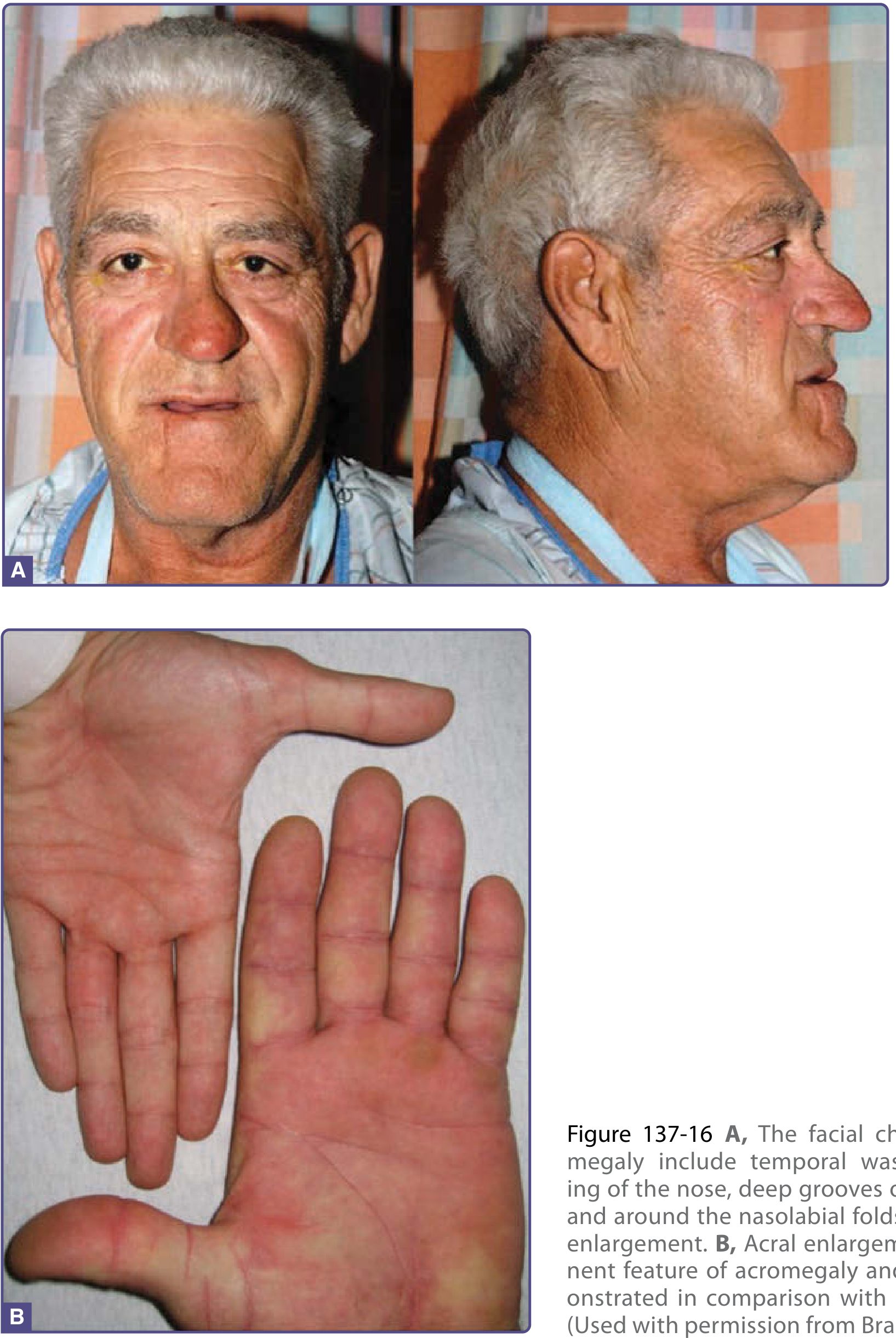
1. Diagnosis and Supporting Features
Diagnosis: Acromegaly
Features in this patient that suggest the diagnosis:
| Clinical Feature | Significance in Acromegaly |
|---|---|
| Large jaw | Prognathism - mandibular overgrowth from excess GH/IGF-1 |
| Large hands (rings no longer fit) | Acral enlargement - soft tissue and bony growth |
| Thick, coarse hair; hirsutism | GH stimulates hair follicles; androgen excess from co-secreting adenoma |
| Amenorrhea / menstrual irregularity | Tumor compression of pituitary causing hyperprolactinemia (25-30% of cases) or hypopituitarism with suppression of LH/FSH; she believes she is in "early menopause" |
| Headaches "for years" | Local mass effect of pituitary adenoma on surrounding structures |
| Arthritis (hip and knees) | GH excess causes arthropathy via cartilage and joint overgrowth |
| Fatigue and weakness | Metabolic effects of chronic GH excess; may also reflect hypopituitarism |
| Slightly enlarged thyroid | Visceromegaly - GH causes enlargement of thyroid, heart, liver, kidneys |
| Displaced apex beat (cardiomegaly) | GH-induced cardiomyopathy and cardiac hypertrophy |
| "Getting fat" | Soft tissue hypertrophy and fluid retention, not true fat gain |
| Age of onset/insidious course | Average diagnosis in 40s; disease present years before diagnosis |
Additional facial and oral signs to look for:
- Frontal bossing - supraorbital ridge hypertrophy causing a prominent, overhanging forehead
- Macroglossia - enlarged tongue, which may impair speech and contribute to sleep apnea; teeth widely spaced due to jaw growth
- Prognathism with dental malocclusion - the lower jaw protrudes beyond the upper, with separation/spacing of the lower teeth (bite change is a classic early clue patients notice)
2. Five Complications of Acromegaly
-
Cardiovascular disease - hypertension, GH-induced cardiomyopathy with left ventricular hypertrophy, diastolic dysfunction, arrhythmias, and accelerated atherosclerosis. This is the leading cause of death.
-
Diabetes mellitus / glucose intolerance - GH antagonizes insulin action; impaired glucose tolerance occurs in up to 50% of patients; frank diabetes develops in many. The displaced apex beat + goitre + amenorrhea in this patient raises suspicion for concurrent metabolic disease.
-
Sleep apnea - both obstructive (macroglossia, enlarged soft tissues of the pharynx) and central types occur; a common and under-recognized complication.
-
Arthropathy - degenerative joint disease, particularly of the large weight-bearing joints (hips, knees - as reported in this patient) and the spine; caused by cartilage overgrowth followed by degradation.
-
Colon polyps and increased cancer risk - acromegaly is associated with an increased incidence of colonic polyps and possibly colorectal cancer; visceromegaly also affects thyroid (goitre as seen here), heart, liver, and prostate (uterine fibroids/leiomyomata in women).
3. Next Step and Investigations
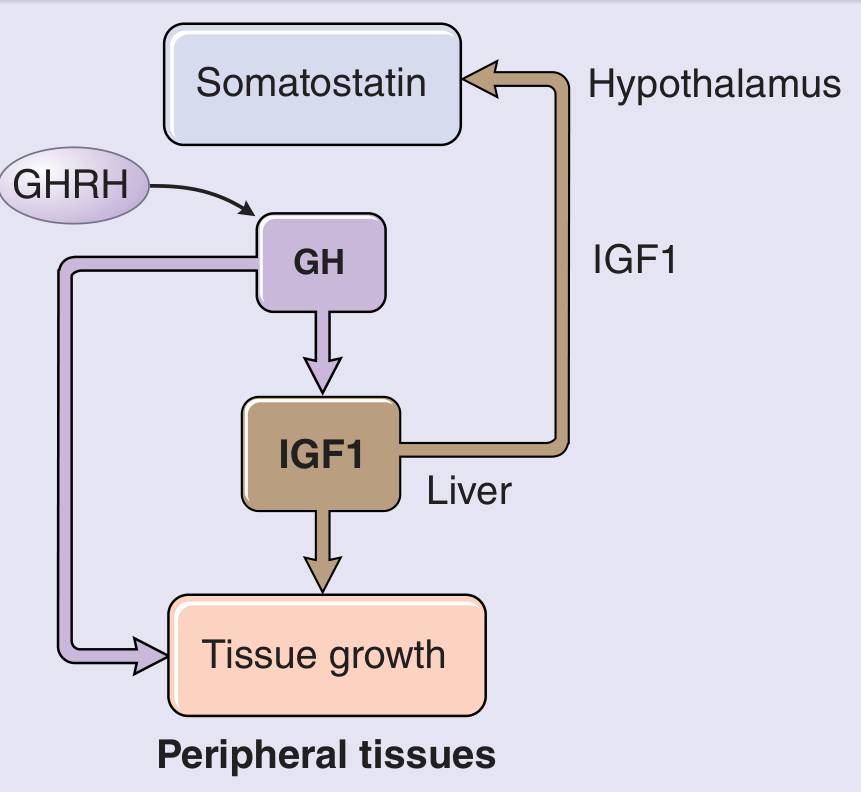
Next step: Measure serum IGF-1 (Insulin-like Growth Factor-1)
"Elevated serum concentration levels of age- and gender-matched serum insulin-like growth factor type 1 (IGF-1) provides the best single test to screen for acromegaly." - Textbook of Family Medicine 9e
Three key investigations and expected findings:
- What you see: Elevated IGF-1 above age- and sex-matched normal range
- GH itself is not reliable due to pulsatile secretion; IGF-1 integrates GH secretion over time
- Give 75 g oral glucose; measure GH at baseline, 1 hour, and 2 hours
- What you see in acromegaly: GH fails to suppress to <1 ng/mL (normal is suppression to <1 ng/mL within 1-2 hours), OR shows a paradoxical rise in GH
- This is the definitive diagnostic test (Goodman & Gilman's; Textbook of Family Medicine 9e)
- What you see: A pituitary adenoma - either microadenoma (<10 mm) or macroadenoma (≥10 mm). Macroadenomas may show suprasellar extension toward the optic chiasm (causing visual field defects)
- If MRI is normal, look for ectopic source (chest/abdomen/pelvis MRI for carcinoid or islet cell tumor)
Additional tests to order:
| Test | Expected Findings |
|---|---|
| Serum prolactin | Elevated in 25-30% (somatomammotropic adenoma or stalk compression) - explains her amenorrhea |
| Fasting glucose / HbA1c | Impaired glucose tolerance or frank diabetes in ~50% |
| Serum phosphate, alkaline phosphatase | Elevated (markers of GH activity) |
| Thyroid function tests | Rule out hypopituitarism-related hypothyroidism |
| Serum calcium | Exclude MEN-1 (parathyroid hyperplasia) |
| Visual field testing (perimetry) | Bitemporal hemianopia from optic chiasm compression by macroadenoma |
| ECG / Echocardiogram | Cardiomegaly, LV hypertrophy, diastolic dysfunction |
| Colonoscopy | Screening for colonic polyps |
4. Best Treatment
- Performed by an experienced neurosurgeon via the transsphenoidal route (through the sphenoid sinus)
- Objectives: remove the adenoma, decompress the optic chiasm if needed, normalize GH and IGF-1 levels, and preserve remaining pituitary function
- Success rate depends on tumor size and surgical expertise: microadenomas have cure rates of 70-90%; macroadenomas have lower cure rates (~40-60%) with higher relapse rates
- Post-operative success is defined as GH <1 ng/mL on OGTT and normalized age-matched IGF-1
- Fatigue, paresthesias, headaches, and soft tissue swelling improve rapidly after successful surgery
5. Other Treatment Options
A. Somatostatin Analogues (SSA) - First-line medical therapy
- Octreotide (Sandostatin LAR) and Lanreotide (Somatuline Depot) - potent, long-acting inhibitors of GH secretion
- Given as once-monthly (octreotide LAR) or biweekly (lanreotide) intramuscular depot injections
- Normalize GH and IGF-1 in ~50% of patients after 18-24 months
- Rapid improvement in fatigue, headaches, paresthesias, and soft tissue swelling
- May shrink tumor in some patients - can be used pre-operatively to improve surgical outcomes
- Side effects: GI symptoms (nausea, bloating, diarrhea), gallstone formation (most significant)
- Hyperhidrosis typically persists even with biochemical normalization
B. GH Receptor Antagonist
- Pegvisomant - a GH receptor antagonist that blocks GH action at target tissues, thereby lowering IGF-1
- FDA-approved; very effective at normalizing IGF-1 (~90% of patients)
- Does NOT directly suppress GH or reduce tumor size - used when SSAs fail or are not tolerated
- Requires liver function monitoring
C. Dopamine Agonists
- Bromocriptine and Cabergoline - suppress GH secretion in some patients
- Less effective than SSAs (normalize GH in only ~10-20%)
- Useful as adjuvant therapy or in patients with co-secretion of prolactin (as in this patient)
- Oral, convenient, and inexpensive
D. Radiation Therapy
- Stereotactic radiosurgery (Gamma Knife) or conventional fractionated radiotherapy
- Reserved for recalcitrant cases: residual/recurrent adenoma after surgery, patients unsuitable for surgery, or failure of medical therapy
- Effect is slow (may take years to normalize GH)
- Risk of hypopituitarism (all pituitary hormone axes), visual damage if tumor is close to optic apparatus, and rare risk of secondary radiation-induced tumors
Treatment Algorithm Summary:
Surgery (transsphenoidal resection) → first line
↓ if residual/recurrent disease
Somatostatin analogues (octreotide/lanreotide) ± dopamine agonists
↓ if SSA resistance or intolerance
Pegvisomant (GH receptor antagonist)
↓ if all above fail
Radiation therapy (Gamma Knife / conventional)
6. Other Conditions with Elevated GH Levels
-
Pituitary Gigantism - excess GH secretion from a pituitary adenoma occurring before epiphyseal plate closure in children/adolescents; results in extraordinary linear growth (tall stature) rather than the bony remodeling seen in acromegaly. GH physiology is identical to acromegaly but the clinical phenotype differs because the epiphyses are still open.
-
Ectopic GH or GHRH-secreting tumors - rare non-pituitary tumors that secrete either GH directly or GHRH (which drives pituitary GH release):
- Carcinoid tumors (lung, bronchus, gastrointestinal tract)
- Pancreatic islet cell tumors
- Small cell lung cancer
- These account for <2% of acromegaly cases but are important to recognize when pituitary MRI is normal
(Other conditions: McCune-Albright syndrome, Carney complex, MEN type 1 - all associated with pituitary GH-secreting adenomas as part of their syndrome)
- Diagnosis is typically delayed by 7-12 years from symptom onset because changes are insidious - this patient's "years" of headaches and progressive body changes reflect this
- Amenorrhea in this woman is explained by hyperprolactinemia (co-secreted in 25-30%) or direct pituitary damage/stalk effect - NOT premature menopause
- The displaced apex beat signals GH-induced cardiomyopathy - the #1 cause of increased mortality
- MRI + IGF-1 + OGTT is the diagnostic triad
- Transsphenoidal surgery is the answer to Q4 (best treatment), with medical/radiation options for residual disease