Discuss thrombogenesis and its complications
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
Finding Sources
Reading File
Finding Sources
I now have all the content needed for a thorough, well-structured answer. Let me compile the comprehensive response.
Thrombogenesis and Its Complications
Thrombogenesis refers to the pathological formation of a thrombus (blood clot) within an intact blood vessel or heart chamber, as distinct from the normal hemostatic response to vascular injury. Understanding it requires examining the triggers, molecular machinery, thrombus morphology, and downstream consequences.
1. Virchow's Triad - The Three Pillars of Thrombosis
The primary abnormalities that lead to thrombosis are captured in Virchow's triad: (1) endothelial injury, (2) stasis or turbulent blood flow, and (3) hypercoagulability. These factors may operate independently or in combination, and each element can feed back to worsen the others.
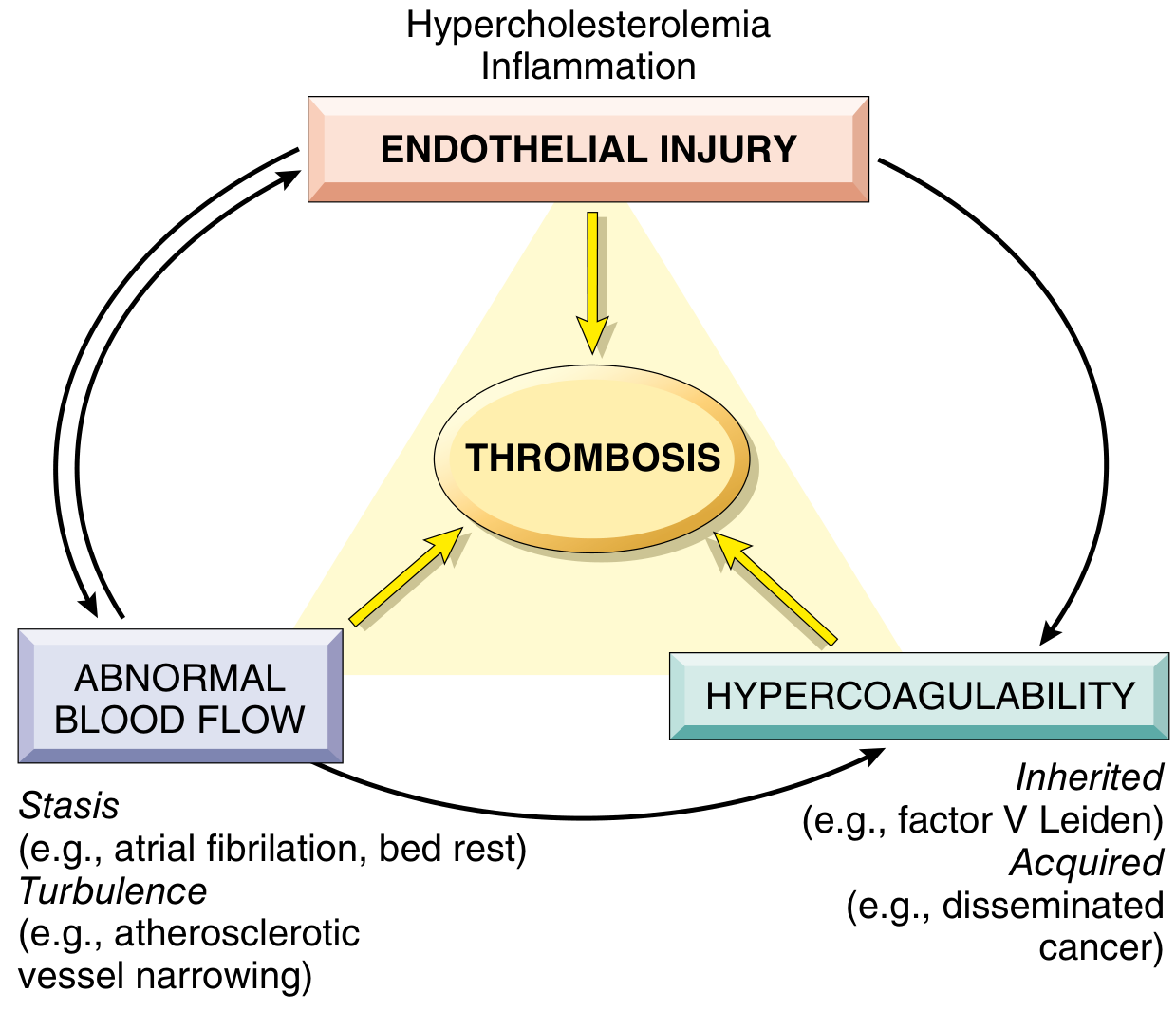
1A. Endothelial Injury
Endothelial injury is the dominant trigger for arterial and cardiac thrombosis, where high-shear blood flow would otherwise prevent clot formation. Cardiac and arterial clots are characteristically platelet-rich, reflecting the centrality of platelet adhesion and activation under these conditions.
Mechanisms of endothelial injury:
- Physical injury, infectious agents, abnormal blood flow
- Inflammatory mediators, hypercholesterolemia, homocysteinemia
- Cigarette smoke toxins
Prothrombotic ("endothelial activation") changes:
- Procoagulant shift: Inflammatory cytokines downregulate thrombomodulin (a key thrombin modulator), protein C, and tissue factor pathway inhibitor (TFPI) - all anticoagulant molecules. The net result is enhanced procoagulant and proinflammatory thrombin activity.
- Antifibrinolytic effects: Activated endothelium secretes plasminogen activator inhibitors (PAIs) and downregulates tissue plasminogen activator (t-PA) expression, inhibiting fibrinolysis.
- Direct exposure of subendothelial matrix: Exposes von Willebrand factor (vWF) and tissue factor (TF), directly initiating platelet plug and coagulation cascade.
1B. Abnormal Blood Flow (Stasis and Turbulence)
Normal laminar flow keeps platelets centrally in the vessel, separated from endothelium by a plasma layer. Turbulence drives arterial/cardiac thrombosis; stasis is the primary driver in venous thrombosis. Both:
- Promote endothelial activation and procoagulant gene expression
- Disrupt laminar flow, bringing platelets into direct endothelial contact
- Prevent washout and dilution of activated clotting factors by fresh blood
- Impede inflow of natural clotting factor inhibitors
Clinical examples:
| Mechanism | Clinical Setting |
|---|---|
| Stasis | Atrial fibrillation, prolonged bed rest, heart failure |
| Turbulence | Atherosclerotic plaque ulceration, aortic aneurysm |
| Stasis (cardiac) | Myocardial infarction with non-contractile myocardium, left ventricular aneurysm |
| Stasis (atrial) | Rheumatic mitral stenosis with left atrial dilation |
| Stasis (microvascular) | Polycythemia vera (hyperviscosity), sickle cell anemia |
1C. Hypercoagulability
Hypercoagulability denotes an abnormally high tendency to clot, caused by alterations in coagulation factors. It is particularly important in venous thrombosis and is divided into primary (genetic) and secondary (acquired) causes.
Table 1: Causes of Hypercoagulability
| Category | Condition | Mechanism/Risk |
|---|---|---|
| Primary (Genetic) | Factor V Leiden | Renders factor V resistant to protein C cleavage; 4-8x risk (heterozygote), ~12x (homozygote); present in ~60% of recurrent DVT patients |
| Prothrombin G20210A mutation | Elevated prothrombin levels; 2-4x risk (heterozygote), 7x (homozygote) | |
| Protein C or S deficiency | Loss of anticoagulant downregulation | |
| Antithrombin III deficiency | Loss of factor inhibition | |
| Secondary (Acquired) | Prolonged bed rest or immobilization | Stasis + reduced fibrinolysis |
| Pregnancy | Venous stasis + elevated procoagulant factors | |
| Oral contraceptive use | Elevated estrogen-driven clotting factor synthesis | |
| Malignancy | Tumor cells release TF and mucin, activating coagulation | |
| Antiphospholipid syndrome | Antibodies activate platelets, endothelium | |
| Disseminated intravascular coagulation (DIC) | Systemic coagulation activation | |
| Heparin-induced thrombocytopenia (HIT) | Antibody-mediated platelet activation | |
| Nephrotic syndrome | Loss of antithrombin III in urine | |
| Older age, obesity, smoking | Multiple interacting mechanisms |
2. Molecular Mechanism of Thrombogenesis
The process unfolds via two parallel, interacting arms: the platelet arm and the coagulation arm.
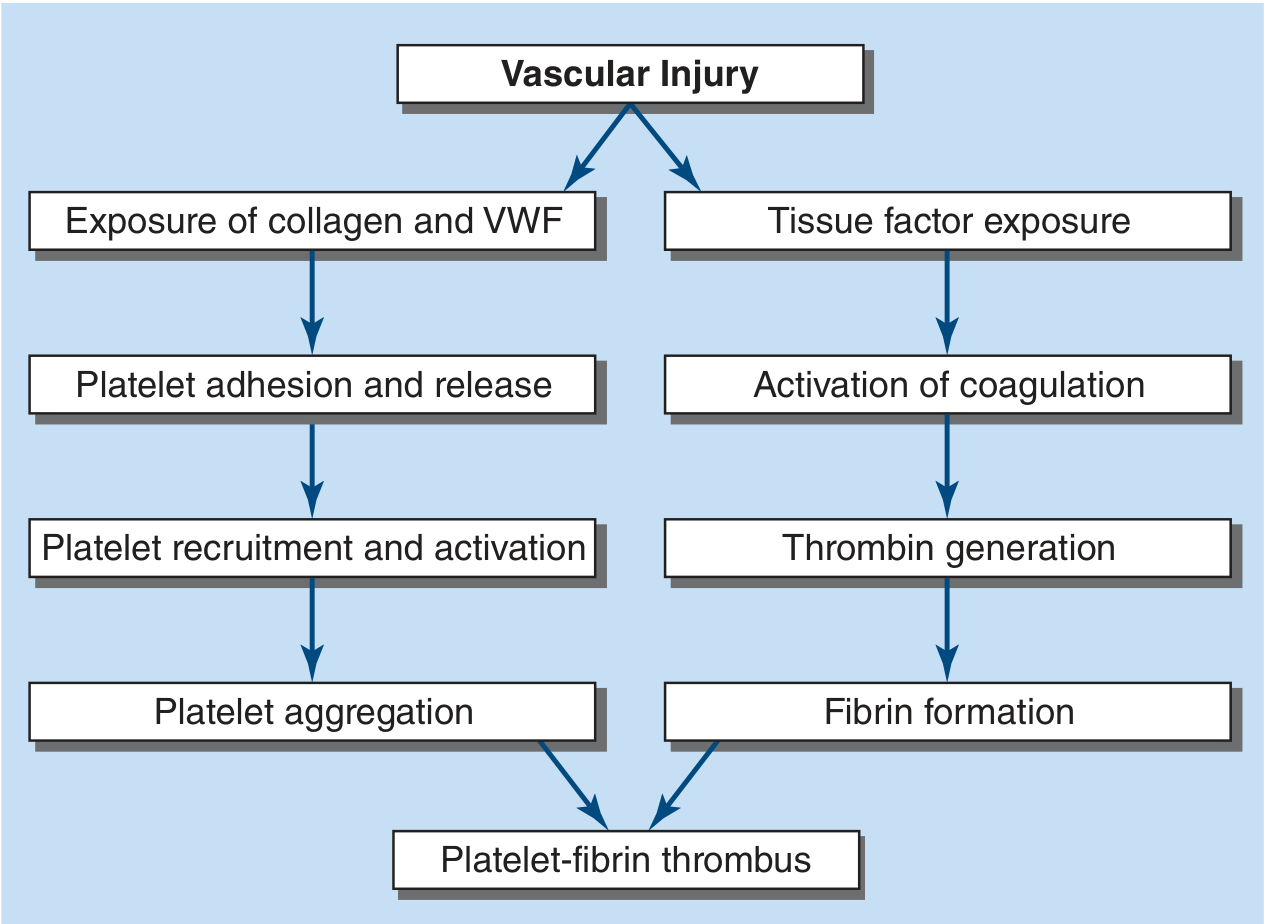
Platelet Arm
- Adhesion: Vascular injury exposes subendothelial collagen and vWF. Platelets adhere via GpIb-vWF interaction and GpVI/alpha2beta1-collagen interaction. In healthy vessels, endothelial NO, prostacyclin (PGI₂), and CD39 (ADPase) keep platelets quiescent.
- Activation and release: Adherent platelets activate, releasing ADP and thromboxane A₂ (TXA₂) - both potent platelet agonists that recruit and activate additional circulating platelets.
- Aggregation: Activation causes a conformational change in GpIIb/IIIa, enabling it to ligate fibrinogen and/or vWF, bridging adjacent platelets into an aggregate.
Coagulation (Fibrin) Arm
- Initiation: Tissue factor (TF) exposed at the site of injury (or carried on bloodborne microparticles and monocytes) binds and activates factor VII, triggering the extrinsic pathway.
- Amplification: Factor VII-TF complex activates factors X and IX; factor Xa generates thrombin (factor IIa). Thrombin amplifies its own production via feedback activation of factors V, VIII, and XI.
- Fibrin formation: Thrombin cleaves soluble fibrinogen into insoluble fibrin monomers. Factor XIIIa (activated by thrombin) cross-links fibrin strands, reinforcing the clot.
- Convergence: Fibrin strands weave the platelet aggregates together to form the definitive platelet-fibrin thrombus.
Key insight: Thrombin serves as the critical amplifier - it is a potent platelet agonist, activates upstream coagulation factors, and converts fibrinogen to fibrin, making it the central node of the entire thrombogenic process.
3. Morphology of Thrombi
- Lines of Zahn: Grossly and microscopically visible pale laminations (platelet and fibrin deposits alternating with red cell-rich layers), forming in flowing blood. Their presence confirms the clot formed ante-mortem (distinguishing from post-mortem clots).
- Post-mortem clots ("chicken fat"): Gelatinous, non-laminated, dark-red dependent layer + yellow upper layer; not adherent to vessel wall.
- Mural thrombi: In heart chambers or aortic lumen; predisposed by arrhythmias, dilated cardiomyopathy, MI, endomyocardial injury, aneurysmal dilation.
- Arterial thrombi: Frequently occlusive; platelet-rich ("white thrombi"); common in coronary, cerebral, and femoral arteries; usually superimposed on ruptured atherosclerotic plaque.
- Venous thrombi (phlebothrombosis): Almost always occlusive; red/stasis thrombi (relatively more enmeshed red cells, fewer platelets); 90% in lower extremity veins; also seen in dural sinuses, portal/hepatic veins.
- Vegetations: Thrombi on heart valves - infected (infective endocarditis) or sterile (non-bacterial thrombotic endocarditis, Libman-Sacks endocarditis in SLE).
4. Fate of the Thrombus
After formation, a thrombus may undergo four possible outcomes:
| Outcome | Description | Clinical Significance |
|---|---|---|
| Propagation | Accumulation of additional platelets and fibrin; thrombus enlarges | Worsens vascular occlusion |
| Embolization | Thrombus fragments dislodge and travel to distant sites | Most dangerous outcome; e.g., PE, stroke |
| Dissolution (fibrinolysis) | t-PA-mediated lysis of fibrin; effective for fresh thrombi | Basis for thrombolytic therapy; ineffective for older cross-linked clots |
| Organization and recanalization | Ingrowth of endothelial cells, smooth muscle cells, and fibroblasts; capillary channels restore flow | Partial restoration of lumen; thrombus ultimately becomes fibrous scar |
Special fate: Occasionally, thrombus centers undergo enzymatic digestion by leukocyte/platelet lysosomal enzymes. If bacteremia occurs, this produces an infected inflammatory mass that can erode the vessel wall and cause a mycotic aneurysm.
5. Complications of Thrombosis
5A. Thromboembolism
The most feared complication. Dislodged thrombus fragments (emboli) travel in the bloodstream to lodge in distant vessels.
- Pulmonary embolism (PE): Venous thrombi (predominantly DVT from lower extremities) embolize to the pulmonary vasculature. Massive PE causes acute right heart failure, hypotension, and death. Saddle embolus straddles the main pulmonary artery bifurcation.
- Systemic arterial embolism: Cardiac mural thrombi (from MI, atrial fibrillation, dilated cardiomyopathy) embolize to brain (stroke), kidneys, spleen, intestines, or limbs.
- Paradoxical embolism: Venous thrombus crosses to arterial circulation via patent foramen ovale or ASD.
5B. Infarction
Arterial thrombi cause downstream ischemia/infarction:
- Myocardial infarction (MI): Coronary artery thrombosis (usually on ruptured atherosclerotic plaque) - the leading cause of STEMI.
- Ischemic stroke: Cerebral arterial thrombosis or cardioembolism.
- Peripheral arterial occlusion: Acute limb ischemia (the "6 P's": pain, pallor, pulselessness, paresthesia, paralysis, poikilothermia).
- Mesenteric ischemia: Occlusion of superior mesenteric artery.
5C. Deep Vein Thrombosis (DVT) and Post-Thrombotic Syndrome
DVT causes acute limb swelling, pain, erythema, and warmth. Chronic venous hypertension after DVT leads to post-thrombotic syndrome: persistent edema, pain, skin hyperpigmentation, lipodermatosclerosis, and venous ulcers.
5D. Disseminated Intravascular Coagulation (DIC)
Pathological systemic activation of the coagulation cascade causes widespread microvascular thrombosis consuming platelets and clotting factors, paradoxically producing simultaneous hemorrhage. Triggered by sepsis, obstetric emergencies (amniotic fluid embolism, abruptio placentae), malignancy, and severe trauma.
5E. Thrombotic Microangiopathy (TMA)
Conditions such as thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) involve microvascular thrombosis causing microangiopathic hemolytic anemia, thrombocytopenia, and end-organ damage (especially kidneys in HUS; neurological in TTP).
5F. Antiphospholipid Syndrome
Arterial and venous thromboses at unusual sites, recurrent miscarriages, thrombocytopenia, mediated by antiphospholipid antibodies (lupus anticoagulant, anti-cardiolipin, anti-beta-2 glycoprotein-I antibodies).
5G. Heparin-Induced Thrombocytopenia (HIT)
Paradoxical complication of heparin therapy where antibodies against platelet factor 4-heparin complexes activate platelets, causing thrombocytopenia and severe arterial/venous thrombosis.
6. Principles of Antithrombotic Therapy
The composition of the thrombus guides treatment choice:
| Thrombus Type | Dominant Component | Primary Treatment |
|---|---|---|
| Arterial (e.g., ACS, stroke) | Platelets | Antiplatelet agents (aspirin, P2Y12 inhibitors, GpIIb/IIIa inhibitors) |
| Venous (DVT/PE) | Fibrin | Anticoagulants (heparin, DOACs, warfarin) |
| Acute massive PE/extensive MI | Both | Fibrinolytic agents (alteplase/t-PA) + anticoagulants |
The addition of low-dose rivaroxaban (factor Xa inhibitor) to dual antiplatelet therapy reduces recurrent ischemic events and stent thrombosis in ACS, highlighting the utility of combined antiplatelet-anticoagulant strategies in high-risk patients.
Key Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Chapter 4: Hemodynamic Disorders, Thromboembolism, and Shock
- Harrison's Principles of Internal Medicine, 22nd Edition (2025) - Chapters 122-123
- Robbins & Kumar Basic Pathology (10th ed.) - Thrombosis
- Mulholland and Greenfield's Surgery, 7e - Venous Thrombogenesis
This is a shared conversation. Sign in to Orris to start your own chat.