Concept of pharmacokinetics
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive material to deliver a thorough answer. Let me compile the full response:
Pharmacokinetics
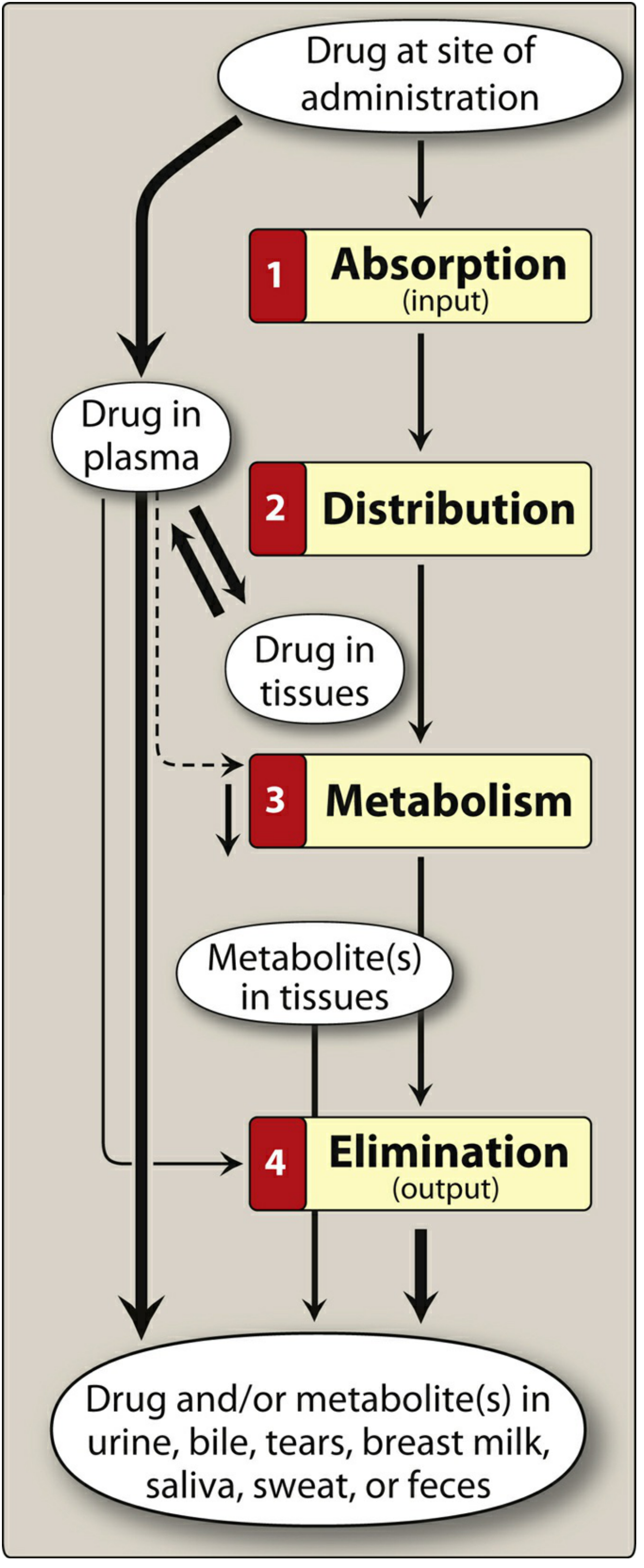
Pharmacokinetics refers to what the body does to a drug - the mathematical and physiological study of how drugs move through the body over time. It encompasses four sequential processes summarized by the acronym ADME: Absorption, Distribution, Metabolism, and Elimination. Clinicians use pharmacokinetic principles to design optimal drug regimens (dose, route, frequency, duration).
1. Absorption
Absorption is the movement of a drug from its site of administration into the bloodstream (plasma).
Factors Affecting Absorption
- Lipid solubility: Lipophilic drugs readily cross cell membranes via passive diffusion. Hydrophilic drugs require protein carriers or pass through aqueous channels.
- Molecular size: Large molecules are absorbed poorly via the oral route.
- Degree of ionization (pKa): The Henderson-Hasselbalch equation governs ionization. Only the non-ionized (uncharged) form of a drug crosses lipid membranes easily. Weak acids are better absorbed in the acidic stomach; weak bases are better absorbed in the alkaline intestine.
- Surface area and blood flow: The small intestine offers the greatest surface area for absorption.
- Formulation: Tablets, liquids, and controlled-release formulations differ in onset and duration.
Routes of Administration
| Route | Key Feature | Example |
|---|---|---|
| Oral | Most common; subject to first-pass metabolism | Acetaminophen, amoxicillin |
| Sublingual | Bypasses first-pass effect; rapid onset | Nitroglycerin |
| IV | 100% bioavailability; immediate effect | Most emergency drugs |
| IM | Moderate absorption | Vaccines, penicillin G |
| Transdermal | Slow, sustained absorption; avoids GI tract | Fentanyl patch, nicotine patch |
| Rectal | Bypasses ~50% of first-pass metabolism | Diazepam suppositories |
| Inhalation | Direct delivery to lung; minimal systemic side effects | Salbutamol, corticosteroids |
Bioavailability (F)
Bioavailability is the fraction of an administered dose that reaches the systemic circulation unchanged. IV administration has an F = 1 (100%). Oral bioavailability is often reduced by:
- First-pass (presystemic) metabolism: After oral absorption, drug passes through the portal vein to the liver, where significant metabolism can occur before reaching systemic circulation. This greatly reduces bioavailability of drugs like lidocaine, morphine, propranolol, and nitroglycerin when taken orally.
2. Distribution
Distribution is the reversible transfer of drug from plasma into interstitial and intracellular fluids and tissues.
Key Determinants
- Plasma protein binding: Albumin is the major carrier protein for acidic drugs (e.g., warfarin, phenytoin); alpha-1-acid glycoprotein binds basic drugs (e.g., propranolol). Only free (unbound) drug is pharmacologically active. Bound drug acts as a reservoir.
- Lipophilicity: Lipophilic drugs freely cross cell membranes; hydrophilic drugs are restricted to extracellular fluid.
- Blood flow: Highly perfused organs (brain, heart, liver, kidney) receive drug first; poorly perfused tissues (fat, bone) equilibrate more slowly.
- Special barriers: The blood-brain barrier (BBB) restricts entry of polar/ionized drugs. The blood-placenta barrier allows lipophilic drugs to cross.
Volume of Distribution (Vd)
Vd is the apparent (hypothetical) volume that would be needed to contain the entire body burden of drug at the same concentration measured in plasma:
Vd = Amount of drug in body / Plasma concentration (C₀)
| Vd Range | Compartment | Example Drug |
|---|---|---|
| ~4 L | Plasma only (highly protein-bound, large MW) | Heparin |
| ~14 L | Extracellular fluid (hydrophilic, small MW) | Aminoglycosides |
| ~42 L | Total body water (lipophilic, small MW) | Ethanol |
| >100 L | Extensive tissue binding | Chloroquine, digoxin |
A large Vd means the drug distributes widely into tissues (low plasma concentration relative to total body load). A small Vd means the drug is confined to plasma/ECF.
3. Metabolism (Biotransformation)
Metabolism converts drugs into more polar, water-soluble metabolites that are more easily excreted. The liver is the primary site (also intestinal wall, lungs, plasma, kidneys).
Phase I Reactions ("Functionalization")
Introduce or expose a polar functional group (-OH, -NH₂, -SH) on the drug molecule. Result: may be active, inactive, or toxic metabolite.
- Cytochrome P450 (CYP) enzymes are the dominant Phase I system
- Key isozymes: CYP3A4 (most abundant, ~50% of drugs), CYP2D6, CYP2C9, CYP2C19, CYP1A2
- CYP inducers (e.g., rifampin, phenytoin, carbamazepine, St. John's wort) increase metabolism → reduced drug effect
- CYP inhibitors (e.g., ketoconazole, clarithromycin, ritonavir, omeprazole) decrease metabolism → drug toxicity risk
- Other reactions: alcohol dehydrogenation (ethanol), esterase hydrolysis (aspirin), amine oxidation (catecholamines)
Phase II Reactions ("Conjugation")
Combine a drug or Phase I metabolite with an endogenous molecule to produce a highly polar conjugate, usually pharmacologically inactive.
| Conjugation Reaction | Endogenous Substrate | Example |
|---|---|---|
| Glucuronidation (most common) | Glucuronic acid | Morphine (to morphine-6-glucuronide, which is MORE potent) |
| Sulfation | Sulfate | Acetaminophen, steroids |
| Acetylation | Acetyl CoA | Isoniazid, sulfonamides |
| Glutathione conjugation | Glutathione | Acetaminophen toxic metabolite (NAPQI) |
| Methylation | Methyl group | Dopamine, norepinephrine |
First-Pass Effect
Orally administered drugs are absorbed from the GI tract and pass through the portal circulation to the liver before reaching systemic circulation. High first-pass metabolism dramatically reduces oral bioavailability.
4. Elimination (Excretion)
Elimination is the irreversible removal of drug (and/or metabolites) from the body.
Renal Elimination (Primary Route)
Three processes operate in the kidney:
- Glomerular filtration: Free (unbound) drug passes passively through the glomerulus. Rate depends on GFR (~120 mL/min/1.73m²). Protein-bound drug is NOT filtered.
- Active tubular secretion: Carrier-mediated transport pumps drug from peritubular capillaries into the tubular lumen (can also secrete protein-bound drug). Examples: organic acid transporters (OAT), organic cation transporters (OCT).
- Passive tubular reabsorption: Lipophilic, non-ionized drugs are reabsorbed back into circulation. Ion trapping exploits this: alkaline urine promotes excretion of weak acids (e.g., alkalinizing urine with sodium bicarbonate promotes aspirin elimination in overdose); acidic urine promotes excretion of weak bases.
Other Excretion Routes
- Biliary/fecal: Large MW drugs and conjugates secreted into bile, reaching intestines. Can undergo enterohepatic recirculation (re-absorption from gut → prolongs drug effect).
- Lungs: Volatile anesthetics, alcohol
- Saliva, sweat, breast milk, tears: Minor routes (but breast milk is clinically relevant for nursing mothers)
5. Key Pharmacokinetic Parameters
Clearance (CL)
The volume of plasma from which drug is completely removed per unit time.
CL = Rate of elimination / Plasma drug concentration
Total body clearance = Renal clearance + Hepatic clearance + Other clearances
Half-Life (t½)
The time required for plasma drug concentration to fall by 50%.
t½ = 0.693 × Vd / CL
- It takes ~4-5 half-lives to reach steady state (during repeated dosing) and ~4-5 half-lives to virtually eliminate a drug from the body.
- Drugs with short half-lives require more frequent dosing.
Steady State (Css)
When the rate of drug input equals the rate of drug elimination. Reached after ~4-5 half-lives of continuous/repeated dosing.
Css = F × Dose / (CL × dosing interval)
Area Under the Curve (AUC)
AUC represents the total drug exposure over time. It reflects bioavailability and is used to calculate clearance.
Zero-Order vs. First-Order Kinetics
| Parameter | First-Order | Zero-Order |
|---|---|---|
| Elimination | Constant fraction eliminated per unit time | Constant amount eliminated per unit time |
| t½ | Constant, independent of dose | Not constant; increases with dose |
| Examples | Most drugs | Aspirin (at high doses), ethanol, phenytoin |
6. Clinical Significance
Understanding pharmacokinetics allows clinicians to:
- Adjust doses in renal or hepatic impairment (reduced clearance)
- Predict drug-drug interactions (CYP inducers/inhibitors)
- Design dosing regimens (loading dose to rapidly achieve therapeutic levels; maintenance dose to sustain steady state)
- Therapeutic Drug Monitoring (TDM): Measure plasma drug concentrations for drugs with narrow therapeutic windows (e.g., digoxin, vancomycin, lithium, phenytoin)
Loading dose = Vd × Target plasma concentration / F Maintenance dose = CL × Target plasma concentration × Dosing interval / F
Sources:
- Lippincott Illustrated Reviews: Pharmacology, Chapter 1 - Pharmacokinetics
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 2 - Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, Metabolism, and Elimination
This is a shared conversation. Sign in to Orris to start your own chat.