Chronic myeloid leukemia
Reading File
Finding Sources
Searching PubMed
"chronic myeloid leukemia" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
I now have all the material needed for a thorough, well-illustrated answer. Let me compile the full response.
Chronic Myeloid Leukemia (CML)
Definition
Chronic myeloid leukemia (CML) is a clonal hematopoietic stem cell (HSC) disorder caused by the reciprocal chromosomal translocation t(9;22)(q34;q11), which fuses the BCR gene on chromosome 22 with the ABL1 tyrosine kinase gene on chromosome 9. This produces the Philadelphia (Ph) chromosome - the shortened chromosome 22 - and the chimeric BCR-ABL1 oncoprotein, a constitutively active tyrosine kinase that drives the disease.
There is no BCR-ABL-negative CML. "Atypical CML" and chronic neutrophilic leukemia are genetically distinct entities.
- Goldman-Cecil Medicine, p. 1938
Molecular Biology
Figure: The Philadelphia chromosome and BCR-ABL1 fusion
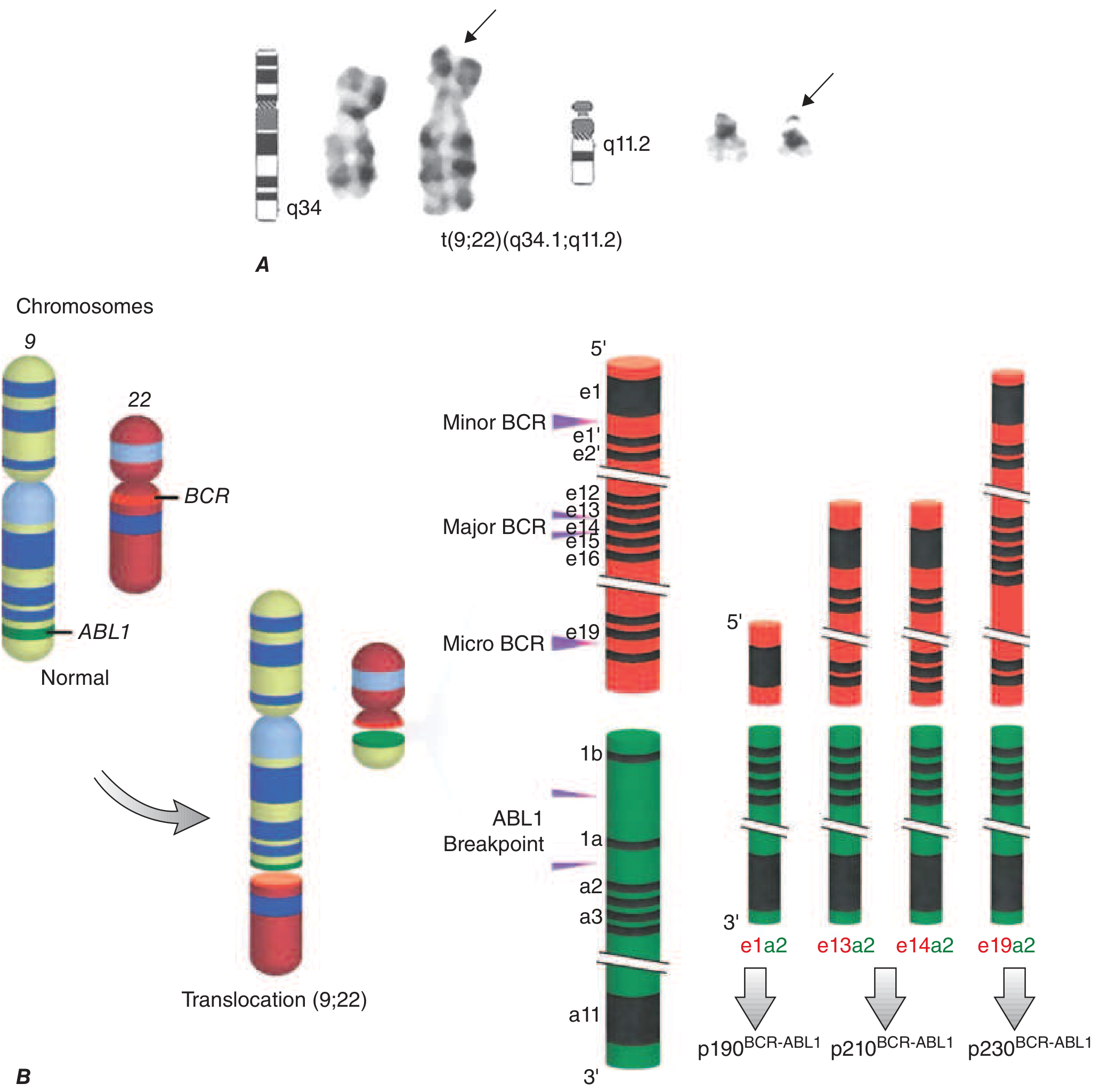
Harrison's Principles of Internal Medicine 22E, Fig. 110-1
The BCR breakpoint determines which oncoprotein is produced:
- p210^BCR-ABL1^ (Major BCR, e13a2/e14a2): Most common in CML (~95%)
- p190^BCR-ABL1^ (Minor BCR, e1a2): Found in ~2/3 of Ph+ ALL, rare in CML
- p230^BCR-ABL1^ (Micro BCR, e19a2): Rare, associated with an indolent course
The BCR moiety contains a dimerization domain that self-associates, constitutively activating the ABL kinase, which then phosphorylates downstream substrates activating RAS, JAK/STAT, PI3K/AKT, and other pro-growth/pro-survival pathways. BCR-ABL preferentially drives proliferation of granulocytic and megakaryocytic progenitors and causes abnormal release of immature granulocytes from marrow.
| Disorder | Mutation | Frequency | Consequence |
|---|---|---|---|
| CML | BCR::ABL fusion | 100% | Constitutive ABL kinase activation |
| PV | JAK2 | >95% | Constitutive JAK2 activation |
| ET | JAK2 / CALR / MPL | Variable | Kinase/receptor activation |
| PMF | JAK2 / CALR / MPL | Variable | Kinase/receptor activation |
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 583
Epidemiology
- Accounts for ~15% of all leukemias
- Annual incidence: ~2 per 100,000; ~9,000 new cases/year in the US
- Median age at diagnosis: 55-65 years (slight male predominance, M:F ~1.6:1)
- Only 3% of patients are under 20 years
- CML prevalence is rising: before TKI therapy, median survival was 3-6 years; with modern TKIs, annual mortality is reduced to ~1-2%, and US prevalence is projected to plateau at ~450,000 by 2040
- No familial associations - not increased in twins or relatives of patients
- Ionizing radiation exposure (nuclear accidents, high-dose RT) does increase risk; peaks 5-10 years post-exposure
- Harrison's Principles of Internal Medicine 22E, p. 878
Disease Phases
CML follows a triphasic natural history:
1. Chronic Phase (CP)
- Asymptomatic accumulation of differentiated myeloid cells in marrow, spleen, and blood
-
75% of cases in the developed world are diagnosed here (often incidentally on routine CBC)
- Peripheral blood shows leukocytosis (often >100,000 cells/μL), with the full granulocytic spectrum: neutrophils, bands, metamyelocytes, myelocytes, eosinophils, basophilia (characteristic)
- Blasts typically <10% in blood
- Platelets often elevated, sometimes markedly
- Symptoms (if any): fatigue, weight loss, left upper quadrant discomfort/early satiety (splenomegaly), anemia
2. Accelerated Phase (AP)
- Increasing anemia and thrombocytopenia, rising basophilia
- Additional cytogenetic abnormalities (e.g., trisomy 8, isochromosome 17q, duplication of Ph chromosome)
- ELN definition: peripheral/BM blasts 15-30%; WHO definition: 10-20%
3. Blast Phase / Blast Crisis (BP)
- Resembles acute leukemia; ELN: blasts ≥30%; WHO: blasts ≥20%
- Myeloid blast crisis in ~70% of cases
- Lymphoid (pre-B cell) blast crisis in ~25% - evidence of pluripotent HSC origin
- Blast crisis is triggered by acquired mutations in transcriptional regulators (e.g., RUNX1, IKZF1)
Without treatment, median survival is ~3 years. About 50% of patients enter an accelerated phase first; the other 50% transform abruptly to blast crisis.
Morphology
Bone marrow: Markedly hypercellular; massively increased maturing granulocytic precursors; increased eosinophils and basophils; dysplastic dwarf megakaryocytes; scattered sea-blue histiocytes (macrophages with wrinkled, green-blue cytoplasm); increased reticulin, but overt fibrosis is rare.
Spleen: Often greatly enlarged due to extensive extramedullary hematopoiesis; may contain infarcts of varying age.
Peripheral blood smear: Basophilia + granulocytosis with neutrophils, bands, metamyelocytes, myelocytes (the full "left-shift"); platelets elevated.
Diagnosis
The diagnosis requires:
- Characteristic blood/BM findings (leukocytosis with granulocytic spectrum, basophilia)
- Detection of the BCR-ABL1 rearrangement by one of:
- Cytogenetics: visualizes the Ph chromosome (90%+ of cases)
- FISH: detects cryptic rearrangements; more sensitive than karyotype
- RT-PCR / quantitative PCR (qPCR): most sensitive; used for monitoring
Important: ~10% of cases have cytogenetically complex/cryptic rearrangements - the Ph chromosome is absent but BCR-ABL1 is detectable by FISH or PCR.
- Washington Manual of Medical Therapeutics, p. 866
Treatment
Tyrosine Kinase Inhibitors (TKIs)
Six FDA-approved oral BCR-ABL1 TKIs exist (as of the current date):
| Agent | Generation | Dose | Notable Toxicities |
|---|---|---|---|
| Imatinib (Gleevec) | 1st | 400 mg daily | Edema, nausea, muscle cramps, rash |
| Dasatinib (Sprycel) | 2nd | 100 mg daily (CP) | Pleural/pericardial effusions, pulmonary HTN |
| Nilotinib (Tasigna) | 2nd | 300 mg BID | QTc prolongation, hyperglycemia, CV events |
| Bosutinib (Bosulif) | 2nd | 400 mg daily | Diarrhea, hepatotoxicity |
| Ponatinib (Iclusig) | 3rd | Variable | Vascular occlusive events, hypertension (via VEGFR inhibition) |
| Asciminib (Scemblix) | 3rd (STAMP) | 40 mg BID (or 200 mg BID for T315I) | Myelosuppression, hypertension, pancreatitis |
- 2nd-gen TKIs are 30-300x more potent than imatinib
- Ponatinib and asciminib are active against the T315I "gatekeeper" mutation (the most resistant kinase domain mutation)
- Asciminib is a STAMP inhibitor (Specifically Targeting the ABL Myristoyl Pocket) - a mechanistically distinct approach from ATP-competitive TKIs
- Harrison's Principles of Internal Medicine 22E, p. 882
Response Milestones (Treatment Monitoring)
Response is monitored by qPCR for BCR-ABL1 transcripts every 3 months. Optimal response milestones:
| Time Point | Target |
|---|---|
| 3 months | Complete hematologic response (CHR) AND BCR-ABL1 ≤10% on IS |
| 6 months | CCyR AND BCR-ABL1 ≤1% on IS |
| 12 months | Major molecular response (MMR) = BCR-ABL1 ≤0.1% on IS |
- CHR: normalization of CBC + absence of splenomegaly
- CCyR: absence of Ph chromosome metaphases on cytogenetic analysis
- MMR (MR3.0): BCR-ABL1 ≤0.1% (IS)
- Deep molecular response (MR4.5): BCR-ABL1 ≤0.0032% - threshold for considering treatment-free remission (TFR)
Failure to meet milestones warrants switching TKIs and mutation testing of the BCR-ABL1 kinase domain.
Treatment-Free Remission (TFR)
Patients who achieve sustained deep molecular response (MR4.5) for ≥2 years may be candidates for stopping TKI therapy. Approximately 40-60% of carefully selected patients maintain remission off therapy.
Blast Phase Management
More challenging - requires combination TKI + intensive chemotherapy + allogeneic hematopoietic stem cell transplantation (HSCT) for eligible patients.
Other Agents
- Omacetaxine mepesuccinate: a protein synthesis inhibitor; option for TKI-resistant disease (including T315I)
- Asciminib (STAMP inhibitor): approved for T315I mutation and after ≥2 prior TKIs
- HSCT: now reserved primarily for blast phase or multiple TKI failures
Survival - TKI Era
Figure: CML survival by era of therapy (MD Anderson experience)
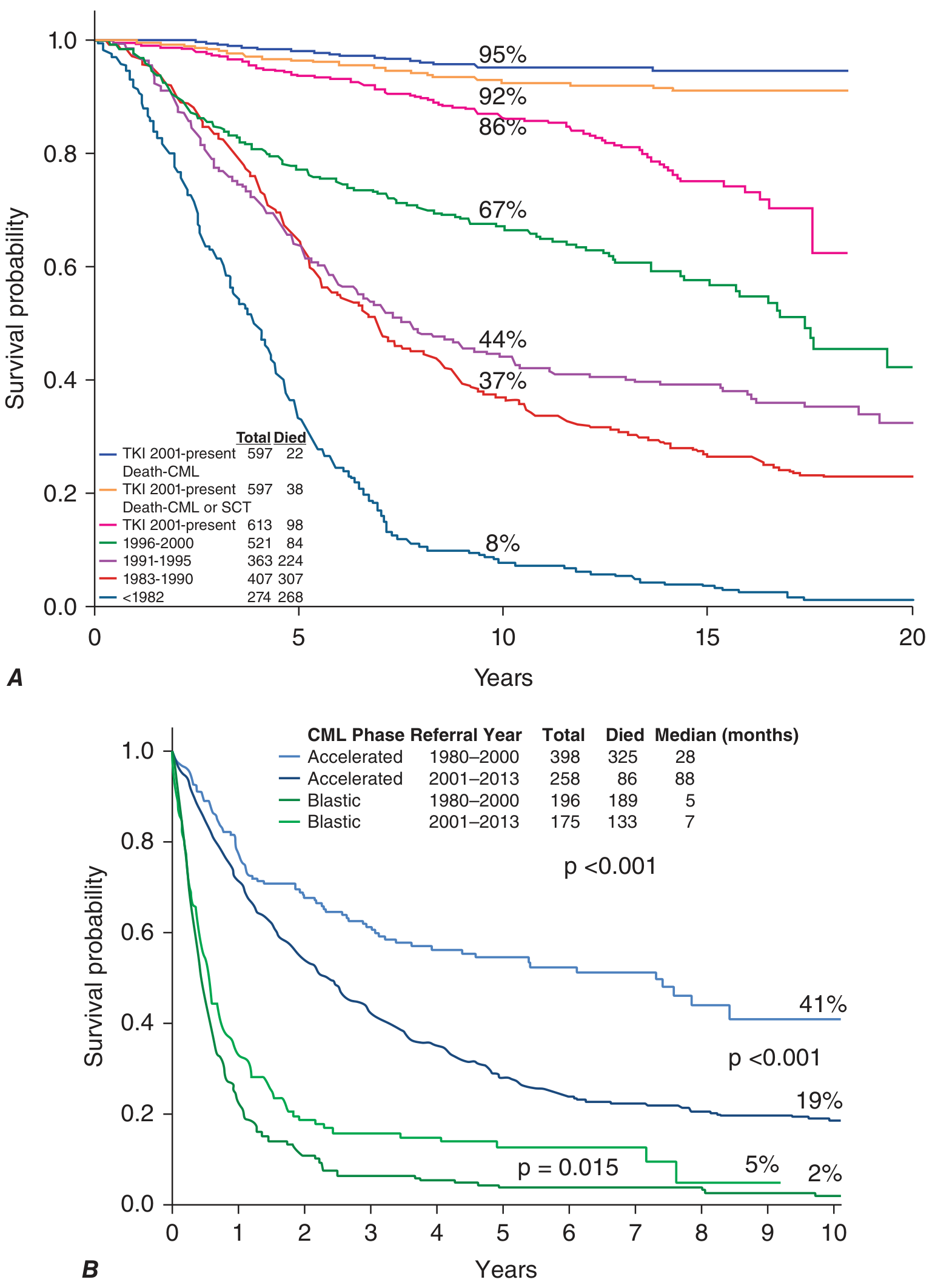
Harrison's Principles of Internal Medicine 22E, Fig. 110-2
- Pre-TKI era (<1982): Only 8% survived 10 years
- TKI era (2001-present): ~95% CML-related survival at 10 years; ~92% accounting for CML/SCT-related deaths; ~86% overall survival
- Accelerated phase: Median survival improved from 28 to 88 months with TKIs
- Blast phase: Still poor prognosis; median survival only 5-7 months, marginally improved with TKIs
Differential Diagnosis
CML must be distinguished from:
- Leukemoid reaction: elevated WBC but with left shift from infection/inflammation; BCR-ABL1 negative; high LAP (leukocyte alkaline phosphatase) score vs. very low in CML
- Other myeloproliferative neoplasms (PV, ET, PMF): BCR-ABL1 negative; JAK2/CALR/MPL mutations
- Atypical CML: morphologically similar but BCR-ABL1 negative; distinct genetics (often SETBP1, CSF3R)
- Chronic neutrophilic leukemia: CSF3R mutations; distinct entity
Recent Evidence (2024-2026)
Three recent systematic reviews are relevant:
-
Ponatinib vs. asciminib after 2nd-generation TKI failure in CP-CML: matching-adjusted indirect comparison showing comparable activity (Garcia-Gutierrez et al., Front Oncol, 2024 - PMID 39634261)
-
TKI toxicity burden: Patient-reported toxicity symptoms during TKI treatment - systematic review and meta-analysis highlighting that symptom burden is significant and often underreported (Smit et al., Support Care Cancer, 2025 - PMID 40317312)
-
TKI + anticoagulant combinations: Safety systematic review for CML patients requiring anticoagulation (Gameil et al., Oncology, 2026 - PMID 41525412)
Summary Table
| Feature | CML |
|---|---|
| Cell of origin | Pluripotent HSC |
| Hallmark mutation | BCR-ABL1 (t(9;22)) - Philadelphia chromosome |
| Oncoprotein | p210^BCR-ABL1^ (most common) |
| Phases | Chronic → Accelerated → Blast |
| Key blood finding | Leukocytosis with full granulocytic spectrum + basophilia |
| Key BM finding | Hypercellularity, sea-blue histiocytes |
| First-line therapy | Any FDA-approved TKI (imatinib, dasatinib, nilotinib, bosutinib) |
| T315I resistance | Ponatinib or asciminib (STAMP) |
| Monitoring | qPCR every 3 months |
| Prognosis (TKI era) | ~95% CML-related 10-year survival in chronic phase |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (p. 583); Harrison's Principles of Internal Medicine 22E (p. 878-882); Goldman-Cecil Medicine (p. 1938); Washington Manual of Medical Therapeutics (p. 866)
This is a shared conversation. Sign in to Orris to start your own chat.