Cardiomyopathy
Reading File
Finding Sources
Searching PubMed
"cardiomyopathy"[MeSH Terms] AND ("classification" OR "management")
Reading File
Reading File
Searching Images
cardiomyopathy types dilated hypertrophic restrictive comparison diagram
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile a thorough, well-organized answer.
Cardiomyopathy
Cardiomyopathies are intrinsic diseases of cardiac muscle — distinct from structural, valvular, ischemic, hypertensive, or congenital causes. There are three major pathophysiologic categories: dilated (90%), hypertrophic, and restrictive (least common), each with distinct mechanics, causes, and management. A fourth category — arrhythmogenic cardiomyopathy — is increasingly recognized.
Echocardiographic Comparison
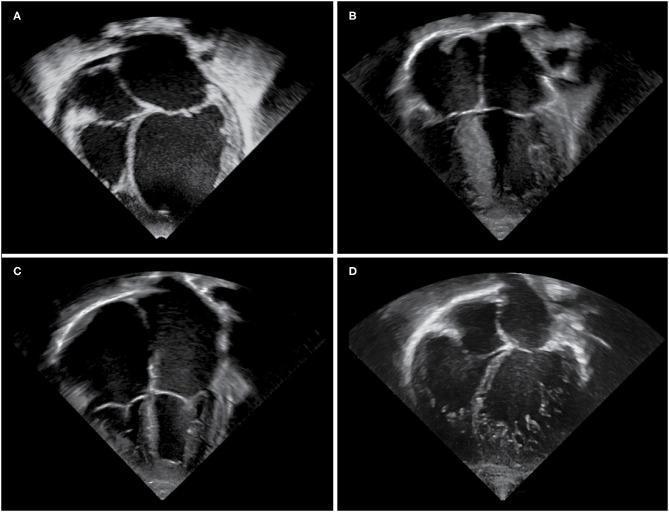
Apical four-chamber echo: A = Dilated CM (globular LV), B = Hypertrophic CM (septal thickening), C = Restrictive CM (biatrial dilation, small ventricles), D = LV Non-compaction (trabeculated endocardium)
Classification by Functional Pattern
| Feature | Dilated | Hypertrophic | Restrictive |
|---|---|---|---|
| LVEF | <40% | 50–80% | 25–50% |
| Primary dysfunction | Systolic (contractile) | Diastolic (relaxation) | Diastolic (compliance) |
| Chamber size | Dilated | Non-dilated (hypertrophied) | Normal/small ventricles, enlarged atria |
| Key causes | Genetic, alcohol, viral, peripartum | Genetic (sarcomere mutations) | Amyloid, radiation fibrosis, sarcoidosis |
1. Dilated Cardiomyopathy (DCM)
Definition & Epidemiology
DCM is characterized by progressive cardiac dilation and systolic dysfunction, usually with concurrent hypertrophy. It is the most common form of nonischemic cardiomyopathy, responsible for ~10,000 deaths and 46,000 hospitalizations per year in the US (incidence ~30/100,000). — Washington Manual of Medical Therapeutics
Pathogenesis
- Genetic (20–50% of cases): >50 genes implicated; autosomal dominant predominant. Key mutations:
- Titin (TTN) truncation — up to 20% of cases; titin is critical for sarcomeric force generation
- Cytoskeletal/sarcolemmal proteins: β-myosin heavy chain, α-myosin heavy chain, cardiac troponin T
- LMNA (nuclear lamins A/C) — often with conduction disease
- Dystrophin (X-linked DCM) — couples cytoskeleton to extracellular matrix
- Desmin — principal intermediate filament protein in cardiac myocytes
- Viral myocarditis: Adenovirus, enterovirus, parvovirus B-19, human herpesvirus 6; coxsackievirus B can progress to chronic DCM
- Toxic: Alcohol and its metabolite acetaldehyde have direct myocardial toxicity; also doxorubicin (anthracycline), cobalt
- Peripartum cardiomyopathy: Occurs in last month of pregnancy or within 5 months postpartum
- Other: Hemochromatosis, sarcoidosis, chronic anemia, idiopathic
Gain-of-function mutations in some of the same sarcomere genes (e.g., MYH7) instead cause hypertrophic cardiomyopathy. — Robbins & Kumar Basic Pathology
Pathology
- Dilated, flabby heart (often >900 g); all four chambers affected
- Microscopy: myocyte hypertrophy, interstitial fibrosis, variable inflammatory infiltrates
- Mural thrombi common → risk of systemic and pulmonary embolism
- Tricuspid and mitral regurgitation due to annular dilation
Clinical Features
- Heart failure symptoms (dyspnea, orthopnea, edema, fatigue)
- Atrial and ventricular arrhythmias in up to 50% → significant SCD risk
- S3 gallop, displaced apical impulse, functional MR murmur
Diagnosis
- Echo/cardiac MRI: Confirms LV dilation and reduced EF
- Endomyocardial biopsy: Not routine; indicated when biopsy result would change management (e.g., new HF <2 weeks + hemodynamic compromise; new HF 2 weeks–3 months + high-grade AVB or ventricular arrhythmias failing usual therapy)
Treatment
- Pharmacologic (same as HFrEF): β-blockers, ARNI (sacubitril/valsartan) or ACEi/ARB, mineralocorticoid receptor antagonists (MRA), SGLT2 inhibitors
- Device therapy: ICD (if EF ≤35% despite optimal therapy), cardiac resynchronization therapy (CRT) for LBBB + reduced EF
- Cardiac transplantation for refractory disease
- Immunosuppression (prednisone, azathioprine) for proven myocarditis has not shown consistent benefit — exception: giant cell myocarditis
2. Hypertrophic Cardiomyopathy (HCM)
Definition & Epidemiology
HCM is the most commonly inherited heart defect, occurring in 1 in 500 individuals (~500,000 in the US). It is the leading cause of sudden cardiac death (SCD) in young people — ~36% of young athletes who die suddenly have probable or definite HCM. — Washington Manual
Pathogenesis
- Autosomal dominant gain-of-function mutations in sarcomere proteins
- Most common mutations: MYBPC3 (myosin binding protein C) and MYH7 (myosin heavy chain 7) — together account for >50% of identified mutations
- Histology: hypertrophied myocytes in disorganized ("whorled") arrangement with interstitial fibrosis — the pathognomonic myocyte disarray
- Results in asymmetric septal hypertrophy (though any segment can be involved)
Pathophysiology
- Diastolic dysfunction: Delayed relaxation and reduced compliance → elevated filling pressures → pulmonary congestion
- LVOT obstruction (~70% at rest or with provocation): due to systolic anterior motion (SAM) of the mitral valve anterior leaflet → worsened by ↑ contractility (exercise), ↓ preload (Valsalva, dehydration), ↓ afterload (vasodilators)
- Myocardial ischemia: Supply-demand mismatch (hypertrophied myocardium + compressed intramural vessels)
- Mitral regurgitation from SAM
Clinical Features
- Exertional dyspnea, angina, syncope, palpitations
- SCD most common ages 10–35, often during or immediately after strenuous exertion
- Murmur: Coarse systolic murmur at left sternal border, louder with Valsalva/standing (↓ preload) and softer with squatting (↑ preload) — distinguishes HCM from aortic stenosis
- Bisferiens pulse (double-peak carotid pulse) in obstruction
- Double or triple apical impulse
Diagnosis
- Echo: LV wall thickness ≥15 mm not explained by loading conditions; SAM; LVOT gradient ≥30 mmHg at rest or with provocation
- ECG: ST-T changes, LVH; apical variant → giant negative T-waves across precordial leads (Yamaguchi pattern)
- Genetic testing recommended for patients and first-degree relatives
Treatment
- Asymptomatic: Activity restriction, avoid volume depletion; genetic counseling
- Symptomatic with obstruction:
- β-blockers (first-line) — reduce HR, improve diastolic filling, reduce obstruction
- Non-dihydropyridine CCBs (verapamil) — alternative to β-blockers
- Mavacamten — novel cardiac myosin inhibitor (FDA approved 2022), reduces contractility and LVOT gradient
- Disopyramide — negative inotrope, added to β-blocker for refractory obstruction
- Avoid: Digoxin, dihydropyridine CCBs, nitrates, diuretics in obstructive HCM (worsen obstruction)
- Septal reduction therapy (surgical myectomy or alcohol septal ablation) for refractory LVOT obstruction
- ICD for SCD prevention in high-risk patients (prior cardiac arrest, sustained VT, family history of SCD, massive LVH ≥30 mm, unexplained syncope, LVEF <50%, abnormal BP response to exercise)
3. Restrictive Cardiomyopathy (RCM)
Definition
RCM results in a stiff, noncompliant myocardium with impaired ventricular filling (diastolic dysfunction), usually with preserved or only mildly reduced systolic function. — Robbins, Cotran & Kumar
Causes
| Category | Examples |
|---|---|
| Infiltrative | Amyloidosis (AL, ATTR), sarcoidosis |
| Storage diseases | Hemochromatosis, Fabry disease, Gaucher disease |
| Endomyocardial | Endomyocardial fibrosis (eosinophilic), Löffler endocarditis |
| Iatrogenic | Radiation-induced fibrosis |
| Idiopathic | — |
Pathophysiology
- Ventricular walls are rigid and noncompliant but systolic contraction relatively preserved
- Massive biatrial dilation from chronically elevated filling pressures (characteristic echo finding)
- Hemodynamics mimic constrictive pericarditis but cannot be relieved by pericardial surgery
Key Clinical Features
- Progressive exertional dyspnea, peripheral edema, ascites
- Elevated JVP with Kussmaul sign (paradoxical rise in JVP with inspiration)
- Distinguishing from constrictive pericarditis is clinically challenging (requires cardiac catheterization ± CMR)
Amyloid Cardiomyopathy (Special Emphasis)
- AL amyloidosis (plasma cell dyscrasia) and ATTR amyloidosis (transthyretin, wild-type or hereditary)
- Echo: "sparkling" myocardium, increased wall thickness, granular texture, small LV cavity, diastolic dysfunction
- CMR: global subendocardial late gadolinium enhancement
- Tafamidis (transthyretin stabilizer) is approved for wild-type and hereditary ATTR cardiomyopathy — reduces mortality and hospitalizations
4. Arrhythmogenic Cardiomyopathy (ACM / ARVC)
- Previously called arrhythmogenic right ventricular cardiomyopathy (ARVC); now recognized to affect both ventricles
- Genetic: Desmosomal protein mutations (PKP2, DSP, DSG2, DSC2, JUP) — autosomal dominant
- Fibro-fatty replacement of myocardium → ventricular arrhythmias, SCD, progressive heart failure
- ECG: epsilon waves, T-wave inversions in V1–V3
- Diagnosis: Task Force Criteria (major/minor from ECG, imaging, histology, genetics, family history)
- Treatment: ICD, antiarrhythmics, activity restriction, transplantation for end-stage disease
Myocarditis (Precursor to DCM)
Myocardial inflammation from infections or immune reactions can progress to DCM. Key forms:
- Viral (most common in US): Lymphocytic infiltrate; coxsackievirus B, parvovirus B-19, HHV-6
- Giant cell myocarditis: Multinucleate giant cells, widespread necrosis, poor prognosis — may respond to immunosuppression
- Hypersensitivity myocarditis: Eosinophil-rich infiltrate, drug reaction
- Chagas myocarditis (T. cruzi): Trypanosomes in myofibers; chronic phase → progressive CHF
Summary: Key Distinguishing Features
| DCM | HCM | RCM | |
|---|---|---|---|
| Ventricular size | Dilated | Normal or small | Normal or small |
| Wall thickness | Normal or thin | Increased (≥15 mm) | Increased (amyloid) or normal |
| EF | Reduced | Preserved/↑ | Preserved |
| Primary problem | Systolic failure | Outflow obstruction / diastolic failure | Diastolic failure (stiff walls) |
| Murmur | Functional MR | Systolic, louder with Valsalva | None characteristic |
| Genetics | Titin, LMNA, DSP | MYBPC3, MYH7 | PKP2 (ACM); hereditary amyloid (ATTR) |
| SCD risk | Moderate (arrhythmias) | High (young athletes) | Variable |
| Treatment anchor | GDMT for HFrEF | β-blockers, mavacamten, septal reduction | Treat underlying cause (e.g., tafamidis for ATTR) |
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; The Washington Manual of Medical Therapeutics; Tintinalli's Emergency Medicine
This is a shared conversation. Sign in to Orris to start your own chat.