Investigation of nephrotic syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
"nephrotic syndrome" AND diagnosis
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the information needed. Here is the comprehensive answer:
Investigation of Nephrotic Syndrome
Nephrotic syndrome is defined as urinary protein excretion >3.5 g/24 hours, serum albumin <3.5 g/dL, hyperlipidemia, and clinically apparent edema. Investigation aims to: (1) confirm the diagnosis, (2) identify the underlying cause (primary vs. secondary), and (3) detect complications.
1. Urine Investigations
Urine Dipstick and Microscopy
- Dipstick typically shows 3+ to 4+ proteinuria
- Urine microscopy: look for oval fat bodies, fatty casts (Maltese cross pattern under polarized light), and lipiduria - hallmarks of nephrotic syndrome
- Hematuria (RBCs or RBC casts) suggests a nephritic component; their presence should prompt consideration of lupus nephritis, MPGN, or IgA nephropathy
Quantification of Proteinuria
- 24-hour urine protein collection: gold standard; nephrotic range = >3.5 g/day in adults (>40 mg/m²/hr in children)
- Spot urine protein:creatinine ratio (PCR): correlates well with 24-hour collection; PCR >3.0 is consistent with nephrotic syndrome. First-morning void sample is most accurate. This is now preferred in clinical practice, especially in children
- Normal values: >2 years old PCR <0.2; 6 months-2 years PCR <0.5
Urine Protein Electrophoresis (UPEP)
- Distinguishes selective (predominantly albumin - suggests MCD) from non-selective proteinuria (mixed - suggests FSGS, membranous nephropathy)
2. Blood Tests
Routine Bloods
| Test | Expected Finding | Purpose |
|---|---|---|
| Serum albumin | <3.5 g/dL (often <2.5 g/dL) | Confirms hypoalbuminemia |
| Serum electrolytes | Hyponatremia (dilutional) | Volume status |
| BUN / Creatinine | Usually normal; may be elevated | Baseline renal function |
| CBC | Elevated Hb/Hct (hemoconcentration) | Assess volume contraction |
| Fasting lipid profile | Elevated total cholesterol, LDL, VLDL; low HDL | Confirms hyperlipidemia |
| Blood glucose + HbA1c | Elevated in diabetic nephropathy | Screen for DKD |
| LFTs | Help exclude hepatic hypoalbuminemia |
Immunological / Serological Screen
These are essential to identify secondary causes:
| Test | Indication |
|---|---|
| ANA, anti-dsDNA, anti-Sm | Lupus nephritis (class V = "membranous lupus") |
| Complement C3 and C4 | Low C3 + C4: SLE, MPGN; Low C3 only: post-streptococcal GN, C3 glomerulopathy |
| Hepatitis B surface antigen (HBsAg), HBeAg | Membranous nephropathy secondary to HBV |
| Hepatitis C antibodies | Associated with MPGN and cryoglobulinemia |
| HIV serology | HIV-associated nephropathy (collapsing FSGS) |
| ANCA (MPO and PR3) | Pauci-immune vasculitis; can have nephrotic-nephritic overlap |
| Anti-GBM antibodies | Goodpasture syndrome |
| Anti-PLA2R antibodies | Primary membranous nephropathy (80% sensitive); a recent 2025 systematic review (PMID: 39820570) confirms its role as a multifaceted biomarker |
| Serum protein electrophoresis (SPEP) + immunofixation | Multiple myeloma, amyloidosis (AL type) |
| Cryoglobulins | Cryoglobulinemic vasculitis/MPGN |
| ASO titre | Post-streptococcal GN |
| VDRL/RPR | Syphilis-associated membranous nephropathy |
In cases with nephritic or nephrotic-nephritic syndrome, a low serum complement strongly suggests immune complex-mediated disease. In pure nephrotic syndrome, complement levels are usually normal. - Goldman-Cecil Medicine
3. Imaging
Renal Ultrasound
- Usually the first-line imaging
- In nephrotic syndrome the kidneys are often normal in size or enlarged (echogenic kidneys suggest chronicity or infiltrative disease)
- Can detect structural abnormalities (polycystic kidney disease, reflux nephropathy), ascites, and obstruction
- Generally non-specific but useful for pre-biopsy assessment
Chest X-Ray
- Can demonstrate pulmonary edema, pleural effusion (from fluid overload)
Doppler Ultrasound / CT Venography
- If renal vein thrombosis is suspected (especially in membranous nephropathy with severe proteinuria >10 g/day and albumin <2 g/dL)
- Pulmonary embolism workup (CTPA) if indicated by symptoms
4. Kidney Biopsy
This is the most definitive investigation for identifying the underlying glomerular disease. It provides:
- Light microscopy (H&E, PAS, silver, Masson trichrome stains)
- Immunofluorescence (IgG, IgA, IgM, C3, C4, C1q deposits)
- Electron microscopy (foot process effacement, deposit location)
When is biopsy indicated?
- All adults with nephrotic syndrome (biopsy changes management in most cases)
- Children: NOT routinely required in first-onset typical nephrotic syndrome (assumed MCD and treated empirically with steroids). Biopsy is indicated in children if:
- Age <1 year or >8 years (lower prior probability of MCD)
- Hematuria, hypertension, or elevated creatinine (suggesting nephritic component)
- Steroid-resistant disease (no remission after 4-8 weeks)
- Steroid-dependent or frequently relapsing
Key biopsy findings:
Minimal Change Disease (MCD):
- Light microscopy: normal glomeruli (hence the name)
- Electron microscopy: widespread podocyte foot process effacement - the hallmark finding
- No immune deposits on IF
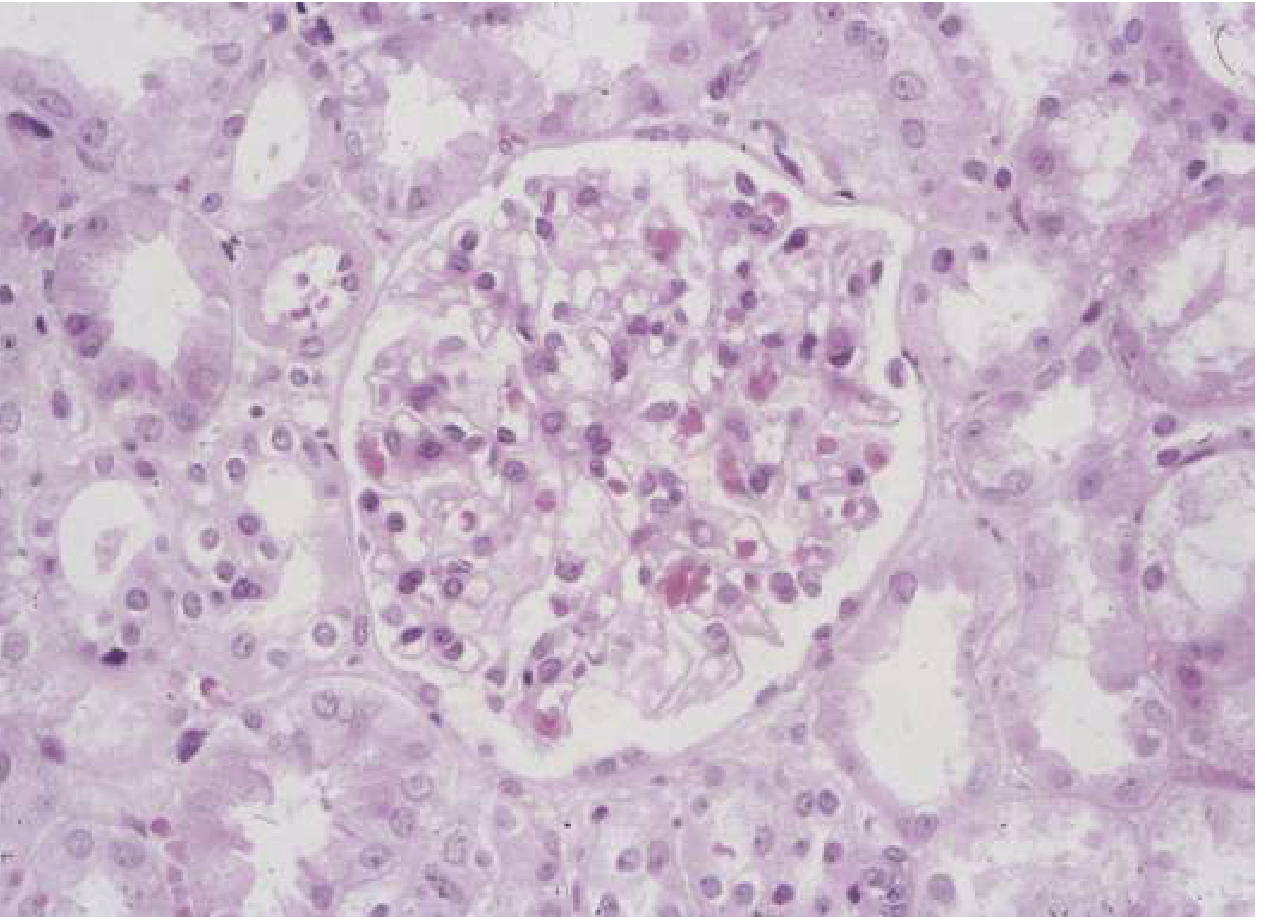
Fig. 107-1: Unremarkable light microscopic appearance of minimal change disease. Capillaries are patent; no hypercellularity. - Goldman-Cecil Medicine
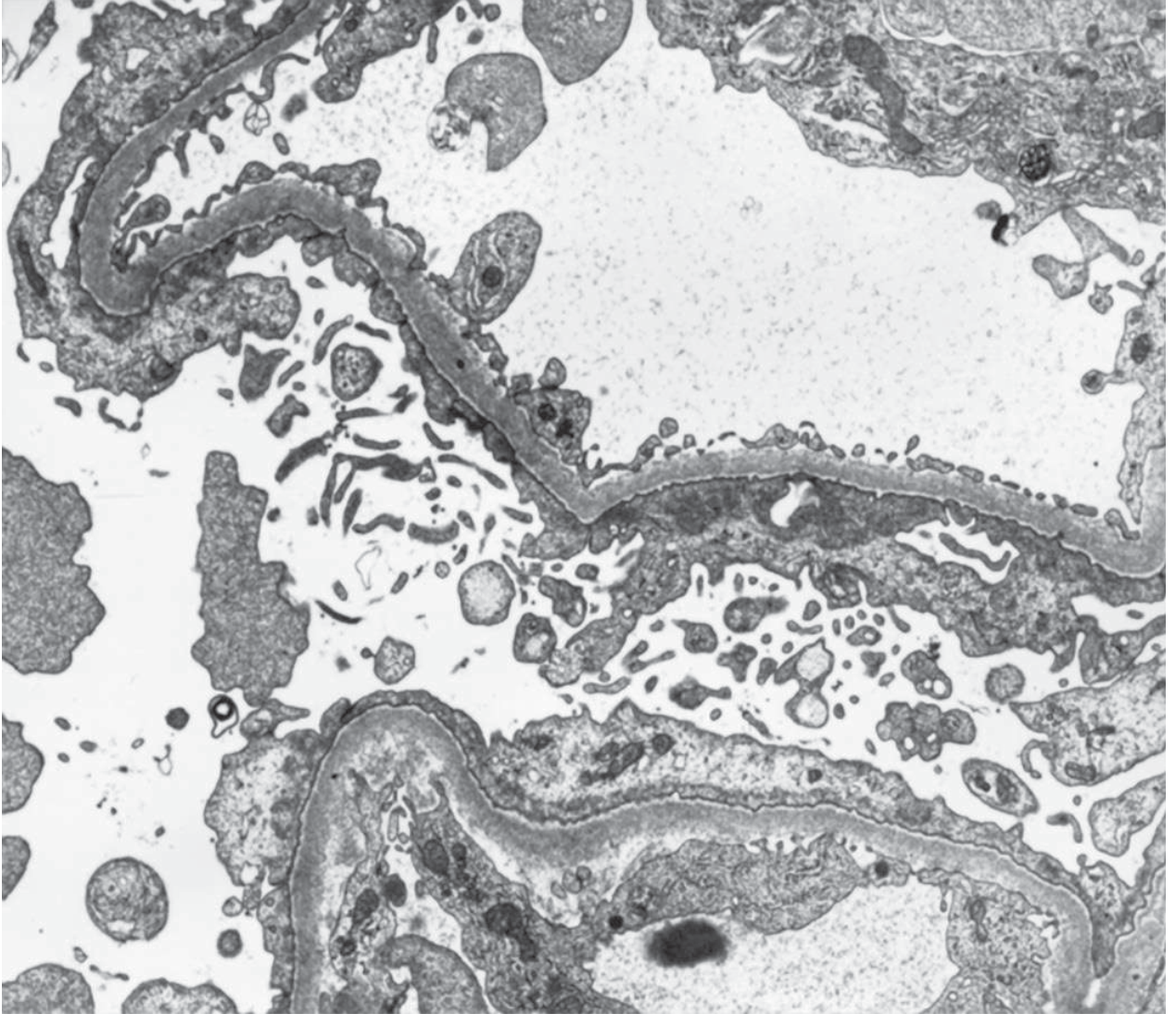
Fig. 107-2: MCD on electron microscopy - widespread effacement of foot processes with microvillus transformation. No electron-dense deposits (×6000). - Goldman-Cecil Medicine
| Disease | Light Microscopy | IF | EM |
|---|---|---|---|
| MCD | Normal | Negative | Diffuse foot process effacement |
| FSGS | Focal, segmental sclerosis | Segmental IgM, C3 | Focal foot process effacement |
| Membranous nephropathy | GBM thickening, "spike and dome" | Granular IgG, C3 (subepithelial) | Subepithelial deposits |
| MPGN | Lobular, double contour GBM | C3 ± IgG (mesangial/subendothelial) | Subendothelial deposits |
| Diabetic nephropathy | Nodular glomerulosclerosis (Kimmelstiel-Wilson) | Often negative | GBM thickening |
| Amyloidosis | Amorphous deposits (Congo red +, apple-green birefringence) | Negative (or monotypic light chains) | Fibrils (8-12 nm diameter) |
5. Additional/Targeted Investigations
| Indication | Test |
|---|---|
| Suspected malignancy (paraneoplastic MN) | CT chest/abdomen/pelvis; tumour markers; age-appropriate cancer screen |
| Drug-induced NS (NSAIDs, gold, penicillamine, lithium) | Medication history; drug levels |
| Amyloidosis | Serum free light chains, bone marrow biopsy, SAP scan (AA amyloid); abdominal fat pad biopsy |
| Sickle cell nephropathy | Haemoglobin electrophoresis |
| Genetic FSGS | NPHS1, NPHS2, APOL1, WT1 gene panel (especially in paediatric or family history) |
| Thrombosis screen | Protein C, Protein S, antithrombin III levels (note: may be spuriously low in active nephrotic syndrome due to urinary loss) |
| Vitamin D deficiency | 25-OH Vitamin D (low due to loss of vitamin D-binding protein) |
| Iron deficiency | Serum iron, transferrin, ferritin (low transferrin in urine) |
| Hypothyroidism | TFTs (thyroid-binding globulin lost in urine) |
6. Summary Algorithm
Suspected Nephrotic Syndrome
↓
Urinalysis + urine PCR (>3.0 = nephrotic range)
↓
Bloods: albumin, lipids, renal function, glucose/HbA1c
↓
Secondary cause screen:
ANA/dsDNA → Lupus
HBV/HCV/HIV → Viral
Complement (C3, C4)
SPEP/immunofixation → Myeloma/amyloid
Anti-PLA2R → Primary membranous
↓
Renal ultrasound
↓
Adults: Kidney biopsy (almost always)
Children (typical NS): Empirical steroids → biopsy if steroid-resistant
Key Complications to Screen For at Presentation
- Hypercoagulability: DVT, pulmonary embolism, renal vein thrombosis - due to urinary loss of antithrombin III, protein C, and protein S. Risk highest in membranous nephropathy (especially with albumin <2 g/dL and proteinuria >10 g/day)
- Infections: Hypogammaglobulinaemia (IgG lost in urine) + T-cell dysfunction - especially pneumococcal peritonitis in children
- AKI: From volume depletion (low oncotic pressure) or acute tubular necrosis
- Metabolic: Vitamin D deficiency, hypothyroidism, iron deficiency anaemia
Sources: Goldman-Cecil Medicine, 26e (p.1252-1254) | National Kidney Foundation Primer on Kidney Diseases, 8e | Rosen's Emergency Medicine | Brenner and Rector's The Kidney, 2-Volume Set | Comprehensive Clinical Nephrology, 7e
Discuss the role of electron microscopy and immunofluroscene in diagnosis of glomerular lesions
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
Finding Sources
I have comprehensive material. Now I can compose the full answer:
Role of Electron Microscopy (EM) and Immunofluorescence (IF) in Diagnosis of Glomerular Lesions
Kidney biopsy is evaluated by three complementary techniques: light microscopy (LM), immunofluorescence (IF), and electron microscopy (EM). While LM provides the overall tissue architecture, IF and EM are essential for determining the pathogenetic mechanism of glomerular injury - particularly the nature, location, and composition of immune deposits. Together these three modalities give a specific diagnosis in most glomerular diseases.
Principles
Immunofluorescence (IF)
IF uses fluorochrome-labeled antibodies directed against immunoglobulins (IgG, IgA, IgM), complement components (C3, C4, C1q), fibrin, and light chains (kappa/lambda) to detect immune deposits in frozen sections of renal tissue. The two critical diagnostic patterns are:
- Granular ("lumpy-bumpy"): Indicates immune complex deposition - either from circulating complexes deposited in the GBM or from in situ immune complex formation. Seen in most glomerulonephritides.
- Linear: Indicates antibody binding to continuously distributed GBM antigens. Classic of anti-GBM disease (Goodpasture syndrome). The antibodies are directed against type IV collagen (α3 chain).
- Negative / No deposits: Seen in MCD, FSGS (primary), diabetic nephropathy, and thrombotic microangiopathies.
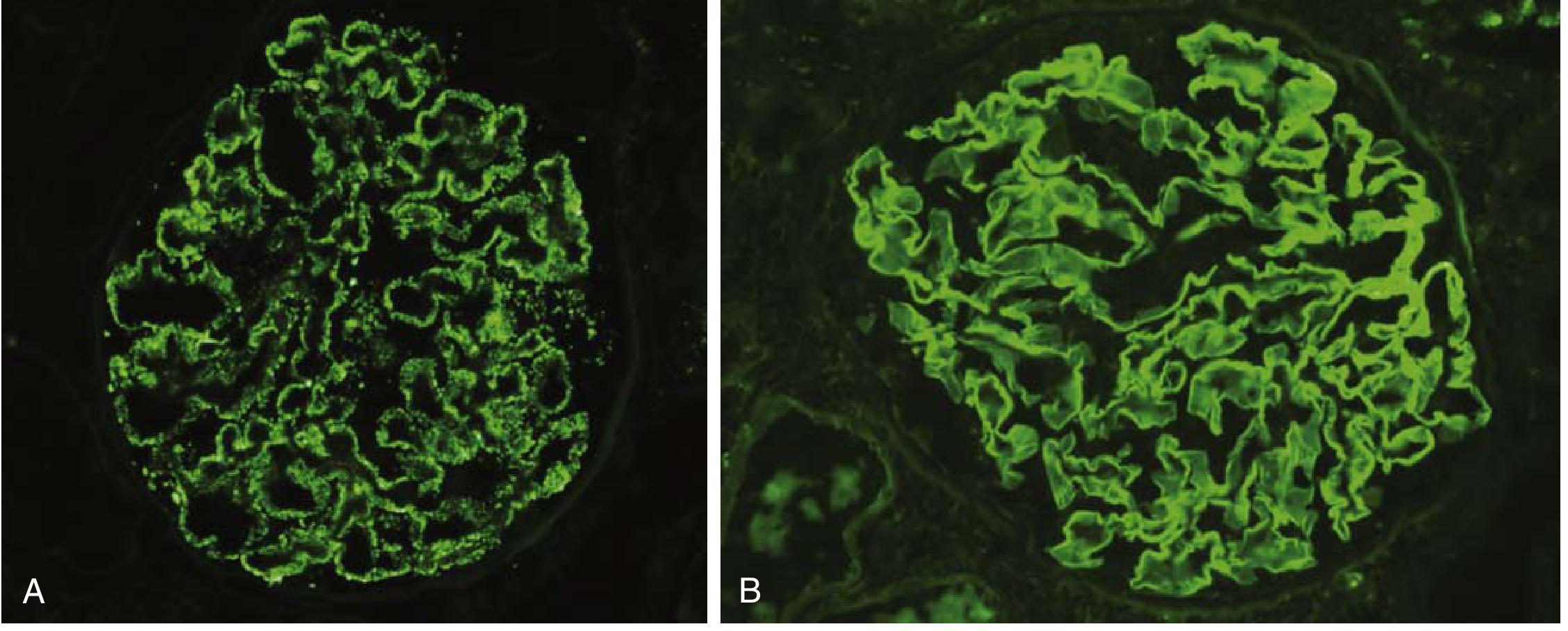
Fig. 12.3 - Patterns of immune complex deposition on immunofluorescence: (A) granular pattern in immune complex GN, (B) linear pattern in anti-GBM disease. - Robbins & Kumar Basic Pathology
Electron Microscopy (EM)
EM resolves ultrastructural detail of the glomerular filtration barrier - the fenestrated endothelium, GBM, and podocyte foot processes - at nanometre resolution. Its key contributions are:
- Localizing immune deposits by their ultrastructural position relative to the GBM:
- Subendothelial (between endothelium and GBM inner lamina): associated with greater inflammation (accessible to complement/leukocytes from blood)
- Subepithelial (between GBM outer lamina and podocyte): often produces proteinuria without overt inflammation - typical of membranous nephropathy
- Intramembranous (within GBM): characteristic of dense deposit disease (DDD)
- Mesangial: associated with IgA nephropathy and early lupus nephritis classes
- Characterizing foot process effacement: The hallmark of all causes of nephrotic syndrome. Foot process effacement (fusion) results from disruption of slit diaphragms
- Identifying specific structures: Fibrils in amyloidosis; tubuloreticular inclusions (TRI) in lupus nephritis; organized microtubular deposits in fibrillary/immunotactoid GN
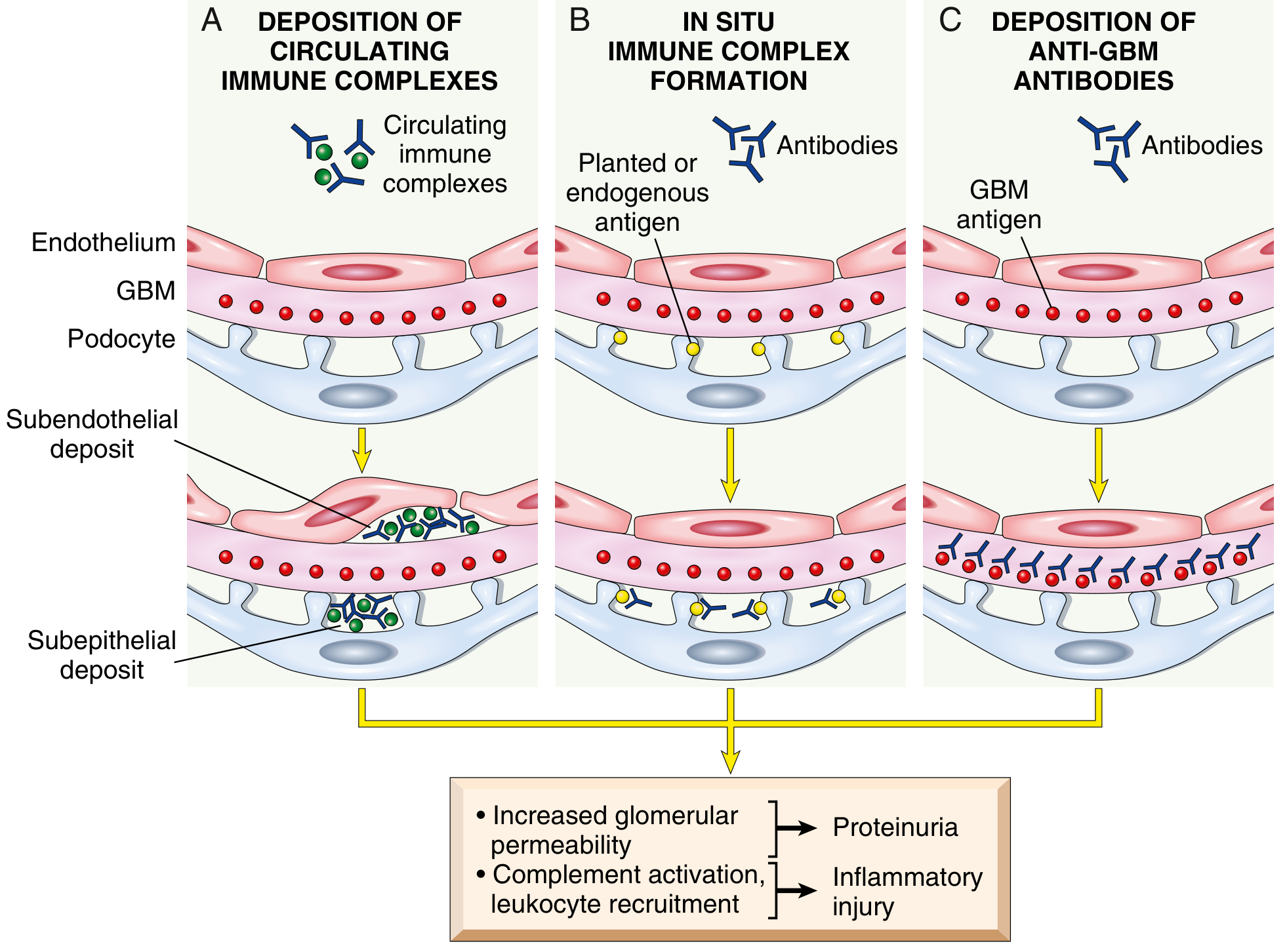
Fig. 12.2 - Mechanisms of glomerular immune injury and their IF patterns. - Robbins & Kumar Basic Pathology
EM and IF Findings in Specific Glomerular Diseases
1. Minimal Change Disease (MCD)
| Finding | |
|---|---|
| IF | Negative - no immune deposits (this is diagnostically critical) |
| EM | Diffuse effacement of podocyte foot processes along virtually all capillary loops + microvillus transformation of podocytes. GBM is structurally normal. No electron-dense deposits |
- The absence of IF deposits distinguishes MCD from all immune complex diseases
- EM foot process effacement is the only abnormality and is the primary pathological finding
- Steroid therapy reverses the foot process changes concomitantly with clinical remission
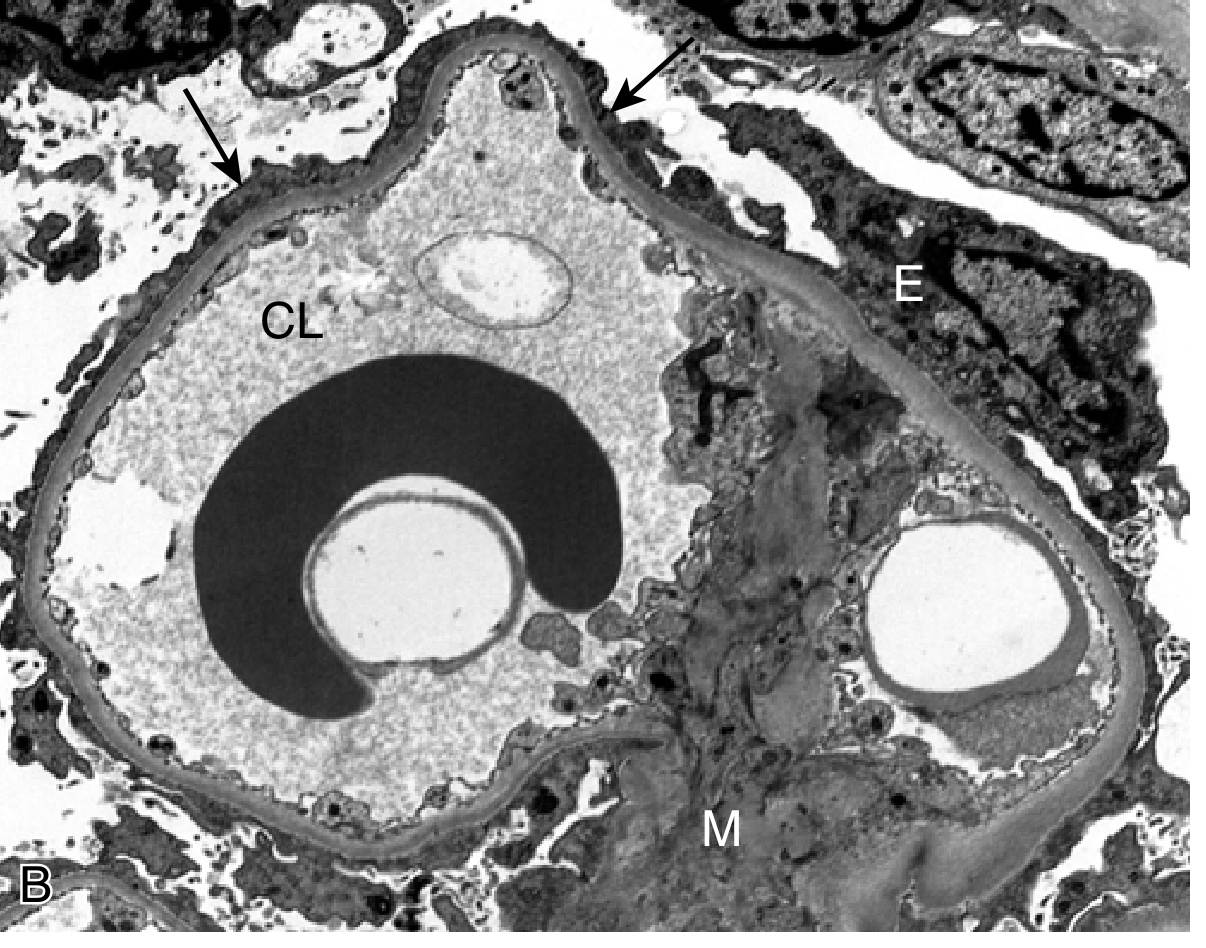
Fig. 12.4B - MCD on EM: widespread foot process effacement (arrows), no deposits. - Robbins & Kumar Basic Pathology
2. Focal Segmental Glomerulosclerosis (FSGS)
| Finding | |
|---|---|
| IF | Nonspecific trapping of IgM and C3 in areas of sclerosis only (not a true immune complex pattern). The non-sclerosed areas are negative |
| EM | Podocyte foot process effacement (more focal than MCD, correlating with degree of sclerosis). No immune-type deposits. Foot process effacement in FSGS typically affects a smaller percentage of capillary loops than in MCD |
- EM helps distinguish FSGS from other causes of focal scarring (e.g., healing post-infectious GN, diabetic nodules) - these may show GBM deposits or structural changes not seen in primary FSGS
- The collapsing variant shows podocyte detachment and retraction on EM
3. Membranous Nephropathy (MN)
| Finding | |
|---|---|
| IF | Granular IgG (predominantly IgG4 in primary MN) and C3 deposits along the GBM in a subepithelial distribution. Uniform, global involvement of all capillary walls. In secondary MN (lupus, HBV), mixed IgG subclasses + mesangial deposits may also be present |
| EM | Subepithelial electron-dense deposits with reactive GBM material forming "spikes" around deposits (the "spike and dome" pattern on silver stain). Extensive podocyte foot process effacement. With disease progression, deposits become surrounded and then incorporated into the GBM ("intramembranous" stage) |
Key features:
- In primary MN: anti-PLA2R antibodies bind to PLA2R antigen on podocytes → in situ subepithelial immune complex formation → granular IF
- EM stages of MN (Ehrenreich-Churg staging): Stage I = subepithelial deposits; Stage II = spikes; Stage III = deposits engulfed by GBM; Stage IV = irregular scarred GBM
4. IgA Nephropathy (Berger's Disease)
| Finding | |
|---|---|
| IF | Dominant or co-dominant mesangial IgA deposition (often with IgG, IgM, and C3). This is the defining diagnostic finding. C3 is frequently co-deposited. C1q is typically absent (distinguishes from lupus) |
| EM | Mesangial and paramesangial electron-dense deposits. Subepithelial "humps" may also be seen in a subset. Foot process effacement is proportional to degree of proteinuria |
- IF is the most critical investigation - IgA in the mesangium is pathognomonic
- Without IF, IgA nephropathy cannot be reliably distinguished from other mesangial GN on LM alone
- Oxford MEST-C classification scores mesangial hypercellularity (M), endocapillary hypercellularity (E), segmental glomerulosclerosis (S), tubular atrophy/interstitial fibrosis (T), and crescents (C)
5. Membranoproliferative Glomerulonephritis (MPGN)
MPGN is now classified by IF into immune complex-mediated and complement-mediated (C3 glomerulopathy) types.
Type I MPGN (immune complex-mediated):
| Finding | |
|---|---|
| IF | Granular IgG, IgM, and C3 in the mesangium and capillary walls (subendothelial distribution). C1q may be present (suggests SLE or cryoglobulinemia) |
| EM | Subendothelial deposits with mesangial deposits. "Tram-track" GBM duplication visible on LM corresponds to mesangial cell interposition between deposits and GBM on EM |
Dense Deposit Disease / C3 Glomerulopathy (complement-mediated):
| Finding | |
|---|---|
| IF | Dominant C3 staining only with little or no immunoglobulin (this distinguishes it from immune complex MPGN). Band-like and coarsely granular C3 in capillary walls and mesangium |
| EM | Intramembranous dense osmiophilic deposits that transform and replace the lamina densa of the GBM, giving it a highly electron-dense, ribbon-like appearance |
6. Poststreptococcal GN (Diffuse Proliferative GN)
| Finding | |
|---|---|
| IF | Granular IgG and C3 deposits in a "starry sky," "mesangial," or "garland" pattern. C3 typically more prominent than IgG |
| EM | Subepithelial "humps" - large, dome-shaped, electron-dense deposits on the outer surface of the GBM between GBM and podocyte. These humps are the pathognomonic ultrastructural finding and correspond to the immune deposits seen in the resolving phase |
7. Anti-GBM Disease (Goodpasture Syndrome)
| Finding | |
|---|---|
| IF | Linear IgG along the GBM (the most striking IF pattern in all of nephrology). This reflects uniform binding of anti-GBM antibodies to the continuously distributed α3(IV) collagen antigen in the GBM |
| EM | No immune-type deposits (since the antibodies bind uniformly to the GBM, they do not form discrete focal deposits). Fibrin in crescents is typically visible. GBM may appear disrupted or fragmented |
- Serum anti-GBM antibodies correlate with IF and confirm the diagnosis without biopsy in many cases
8. Lupus Nephritis
One of the most complex IF/EM patterns - "full house" immunofluorescence is characteristic.
| Finding | |
|---|---|
| IF | "Full house" pattern: IgG, IgA, IgM, C3, C4, C1q all positive (C1q positivity reflects activation of the classical complement pathway by nuclear antigen-antibody complexes). This distinguishes lupus from most other GN |
| EM | Deposits in multiple locations simultaneously: mesangial, subendothelial, subepithelial (reflecting the ISN/RPS class). Class IV (diffuse) shows massive subendothelial deposits forming "wire loops" on LM. Tubuloreticular inclusions (TRI) - parallel tubules within dilated endoplasmic reticulum of endothelial cells, induced by interferon-α - are highly suggestive of lupus nephritis |
9. Diabetic Nephropathy (Diabetic Kidney Disease)
| Finding | |
|---|---|
| IF | Negative (no immune deposits). Linear IgG and albumin staining along the GBM may be seen but reflects non-specific trapping, not true immune complex deposition |
| EM | GBM thickening (earliest finding, detectable before clinical proteinuria). Mesangial matrix expansion without discrete deposits. Foot process effacement proportional to degree of proteinuria |
10. Amyloidosis
| Finding | |
|---|---|
| IF | Monoclonal light chains (kappa or lambda) in AL amyloid. SAA protein in AA amyloid. Staining is typically patchy and may be weak |
| EM | Randomly arranged non-branching fibrils, 8-12 nm in diameter, in the mesangium and GBM. This is the definitive diagnostic finding and distinguishes amyloid from other glomerular deposits |
Summary Table: EM and IF Patterns by Disease
| Disease | IF Pattern | Deposit Location (EM) | EM Key Feature |
|---|---|---|---|
| MCD | Negative | None | Diffuse foot process effacement |
| FSGS | IgM, C3 (nonspecific, in sclerotic areas) | None | Focal foot process effacement |
| Membranous GN | Granular IgG (IgG4), C3 - subepithelial | Subepithelial | "Spike and dome" pattern |
| IgA nephropathy | Dominant mesangial IgA ± C3 | Mesangial | Mesangial/paramesangial deposits |
| MPGN type I | Granular IgG, IgM, C3 | Subendothelial + mesangial | Tram-track GBM |
| Dense deposit disease | C3 only (dominant) | Intramembranous | Dense ribbon in lamina densa |
| Post-streptococcal GN | Granular IgG, C3 | Subepithelial humps | Large dome-shaped humps |
| Anti-GBM | Linear IgG | No discrete deposits | GBM disruption, crescents |
| Lupus nephritis | "Full house" IgG/A/M + C3/C4/C1q | Mesangial + subendo/subepithelial | TRI in endothelium |
| Diabetic nephropathy | Negative | None | GBM thickening, mesangial expansion |
| Amyloidosis | Monotypic light chains (AL) | Mesangial + GBM | Fibrils 8-12 nm, random orientation |
Clinical Significance
- IF is often the single most discriminating test in biopsy interpretation - the IgA predominance in IgA nephropathy, the linear IgG of anti-GBM, and the full house of lupus cannot be inferred from LM alone
- EM confirms and characterizes what IF localizes - it identifies the precise substructural position of deposits, characterizes their internal architecture (fibrils vs. amorphous), and provides a complete picture of podocyte health
- In diseases where IF and LM are both negative (MCD, FSGS, diabetic nephropathy), EM of the foot processes is the only modality that reveals the pathological change driving proteinuria
- Together with LM, all three modalities provide a definitive pathological diagnosis guiding immunosuppressive treatment, prognosis, and genetic counseling
Sources: Robbins & Kumar Basic Pathology (pp. 502-530) | Goldman-Cecil Medicine, 26e (pp. 1252-1259) | National Kidney Foundation Primer on Kidney Diseases, 8e | Comprehensive Clinical Nephrology, 7e | Brenner and Rector's The Kidney
Discuss chemical pathology in case of acute renal failure.discuss pathophysiology of chronic renal failure
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
I have all the content needed for a thorough answer. Here is the comprehensive response:
Part 1: Chemical Pathology in Acute Renal Failure (Acute Kidney Injury)
Acute kidney injury (AKI) is defined as an abrupt decline in GFR over hours to days. The resulting failure of excretory, regulatory, and endocrine functions produces a characteristic set of biochemical derangements.
A. Azotaemia (Elevated BUN and Creatinine)
The most fundamental biochemical marker of AKI is a rising serum creatinine and blood urea nitrogen (BUN).
- Serum creatinine: The most widely used marker of renal function. In AKI, it rises due to decreased GFR (reduced filtration of creatinine) and increased muscle breakdown. Important caveat: fluid resuscitation can artificially lower creatinine by dilution; diuresis can raise it.
- BUN: A product of protein catabolism. Normal BUN:creatinine ratio is ~10:1.
- BUN:Cr ratio >15-20 → suggests prerenal azotaemia (volume depletion, effective underfilling, GI bleeding, hypercatabolic states, or steroid use)
- BUN:Cr ratio ~10 → suggests intrinsic renal (tubular) injury (ATN) where tubular function is damaged and BUN cannot be differentially reabsorbed
- Ratio may be spuriously low in malnutrition, liver disease, or low protein intake
- BUN goal for RRT: Keep BUN <100 mg/dL (37.7 mmol/L), though no absolute threshold
B. Sodium and Water Imbalance
- Hyponatremia is common in AKI, primarily dilutional, due to impaired free water excretion from the failed tubules
- Oliguria (<400 mL/day) is the hallmark of severe AKI - reduced GFR limits the ability to excrete filtered water
- Fluid overload/oedema arises from inability to excrete sodium and water, especially if IV fluids are continued
Fractional Excretion of Sodium (FENa)
A critically important biochemical discriminator in the workup of AKI:
$$FENa = \frac{Urine_{Na} / Plasma_{Na}}{Urine_{Cr} / Plasma_{Cr}} \times 100$$
| FENa | Interpretation |
|---|---|
| <1% | Prerenal azotemia (tubules avidly reabsorbing Na; intact tubular function) |
| >1-2% | Intrinsic AKI/ATN (tubular damage; unable to conserve Na) |
| <1% but intrinsic | Seen in contrast nephropathy, hepatorenal syndrome, early GN, myoglobinuria |
Fractional Excretion of Urea (FEurea) is more useful when diuretics have been given (diuretics falsely raise FENa):
- FEurea <35% → prerenal; >50% → intrinsic
C. Metabolic Acidosis
This is one of the most life-threatening chemical changes in AKI.
- The kidney normally excretes ~50-70 mEq of H⁺/day as NH₄⁺ and titratable acid
- In AKI, this excretion fails, leading to H⁺ retention
- Early AKI: Primarily hyperchloraemic metabolic acidosis (normal anion gap) - Cl⁻ accumulates as NH₄⁺ excretion falls
- Later/severe AKI: High anion gap metabolic acidosis develops as unmeasured anions accumulate (phosphate, sulfate, urate, organic acids). 50% of AKI in critical illness has a normal anion gap; early in AKI, hyperchloremia is the principal source of acidosis; subsequently, 50-60% is caused by unmeasured anions and up to 30% is associated with hyperphosphatemia.
- Indications for RRT: Metabolic acidosis with pH <7.2 is an indication for urgent renal replacement therapy (RRT)
D. Hyperkalaemia
This is the most immediately life-threatening electrolyte abnormality in AKI.
- Potassium is primarily excreted by the kidneys; in AKI, filtered load decreases and tubular secretion fails
- Compounded by:
- Acidosis: H⁺/K⁺ exchange shifts K⁺ out of cells (~K⁺ rises 0.6 mEq/L per 0.1 fall in pH)
- Catabolism/cell lysis: Rhabdomyolysis, haemolysis, tissue necrosis release intracellular K⁺
- Inadequate excretion: Oliguria or anuria prevents K⁺ clearance
- Serum K⁺ >6.5 mEq/L with ECG changes is an emergency indication for dialysis
- ECG changes of hyperkalaemia: peaked T waves → prolonged PR → wide QRS → sine wave → VF
E. Hyponatraemia and Hyperphosphataemia
- Hyperphosphataemia: Phosphate (normally excreted by the kidney) accumulates in AKI. Contributes to metabolic acidosis (~30% of the anion excess in renal acidosis)
- Hypocalcaemia: Secondary to hyperphosphataemia (Ca × PO₄ product precipitation) and impaired renal conversion of 25-OH Vitamin D to active 1,25-(OH)₂ Vitamin D. Hypocalcaemia worsens the cardiac toxicity of hyperkalaemia
F. Hyperuricaemia
- Uric acid (end product of purine metabolism) is normally excreted by the kidney
- In AKI, uric acid accumulates - can worsen tubular injury (urate crystal deposition) and is especially severe in tumour lysis syndrome
G. Urine Chemistry Findings in AKI
| Parameter | Prerenal AKI | Intrinsic (ATN) |
|---|---|---|
| Urine Na⁺ | <20 mEq/L | >40 mEq/L |
| Urine osmolality | >500 mOsm/kg | <350 mOsm/kg |
| FENa | <1% | >1% |
| Urine SG | >1.020 | ~1.010 (isosthenuria) |
| Urine sediment | Hyaline casts | Muddy brown granular casts (ATN hallmark) |
H. Biomarkers of AKI (Beyond Creatinine)
| Biomarker | Source | Significance |
|---|---|---|
| NGAL (Neutrophil gelatinase-associated lipocalin) | Proximal tubule (urine/serum) | Rises within 2h of injury; precedes creatinine rise |
| KIM-1 (Kidney injury molecule-1) | Proximal tubule | Tubular injury marker |
| Cystatin C | All nucleated cells; filtered freely | Earlier marker than creatinine for GFR decline |
| TIMP-2 × IGFBP-7 | Tubular cells | Cell cycle arrest markers; predict AKI development |
| α1-microglobulin | Proximal tubule reabsorption | AUC 0.86-0.88 for predicting need for RRT; useful for proximal tubular dysfunction |
I. Summary - AEIOU Indications for Urgent Dialysis in AKI
| A | Acidosis (metabolic, pH <7.2, refractory) |
| E | Electrolytes (K⁺ >6.5 mEq/L, or Na⁺ <115 or >165 mEq/L) |
| I | Intoxication (dialyzable toxins: methanol, ethylene glycol, salicylates, lithium) |
| O | Overload (pulmonary oedema, fluid overload refractory to diuretics) |
| U | Uraemia (pericarditis, encephalopathy, bleeding, BUN trending to >100 mg/dL) |
Part 2: Pathophysiology of Chronic Renal Failure (CKD)
Chronic kidney disease (CKD) is defined as kidney damage or GFR <60 mL/min/1.73 m² for >3 months. Its pathophysiology can be understood at three levels: (1) mechanisms of initial nephron loss, (2) mechanisms of progressive loss (the final common pathway), and (3) systemic consequences of reduced nephron mass.
A. Causes of Initial Nephron Loss
The major causes in decreasing frequency are:
- Diabetic kidney disease (DKD) - most common in developed countries
- Hypertensive nephrosclerosis
- Glomerulonephritis (primary and secondary)
- Polycystic kidney disease
- Recurrent pyelonephritis/obstructive nephropathy
- Amyloidosis, sickle cell nephropathy, others
B. The Intact Nephron Hypothesis and Single Nephron Hyperfiltration
The central concept in CKD progression is the "intact nephron hypothesis" (Bricker) - surviving nephrons maintain overall homeostasis through adaptive compensatory hypertrophy and hyperfiltration.
Mechanism of hyperfiltration:
- Loss of nephrons → reduction in total renal mass
- Surviving nephrons undergo hypertrophy (glomerular enlargement)
- Increased afferent arteriolar vasodilatation (relatively more than efferent vasoconstriction) → increased intraglomerular hydrostatic pressure
- GFR per nephron rises (single-nephron hyperfiltration)
- This is initially adaptive (maintains solute excretion) but is ultimately maladaptive:
- Increased transcapillary filtration pressure damages glomerular capillary endothelium
- Promotes proteinuria (disruption of size and charge selectivity barriers)
- Activates TGF-β → mesangial expansion → glomerulosclerosis
- Loss of additional nephrons → more hyperfiltration in remaining nephrons → progressive loss → end-stage kidney disease (ESKD)
This is why ACE inhibitors and ARBs (which reduce efferent resistance and lower intraglomerular pressure) and SGLT2 inhibitors (which reduce afferent vasodilation) slow progression - they reduce single-nephron hyperfiltration at the cost of a modest initial GFR decline, but slow the rate of nephron loss long-term.
C. Proteinuria as a Driver of Progression
- Glomerular hypertension increases protein leak across the damaged filtration barrier
- Proteinuria itself is nephrotoxic: filtered proteins (especially albumin, transferrin, complement) are taken up by proximal tubular cells, activating NF-κB, releasing cytokines (MCP-1, RANTES), and recruiting interstitial macrophages and T cells
- This drives tubulointerstitial inflammation and fibrosis - the final common pathway of CKD regardless of initial aetiology
- Hypertension accelerates this process by further raising intraglomerular pressure
D. Tubulo-interstitial Fibrosis - The Final Common Pathway
Regardless of primary cause (glomerular, vascular, or tubulointerstitial), progressive fibrosis is the unifying mechanism:
- TGF-β1 upregulation - the master fibrogenic cytokine
- Epithelial-to-mesenchymal transition (EMT): tubular epithelial cells acquire fibroblast phenotype
- Myofibroblast activation: produce collagen I, III, IV → replace functional parenchyma with scar
- Complement activation: C3a/C5a released locally amplify inflammation
- Renin-angiotensin system (RAS) activation: angiotensin II is directly fibrogenic (stimulates TGF-β, MCP-1) in addition to causing hypertension
The result is progressive nephron dropout, tubular atrophy, interstitial fibrosis, and arteriosclerosis.
E. Systemic Consequences of Reduced GFR - The Uraemic Syndrome
As GFR falls, retained solutes accumulate. These are classified as uraemic toxins: urea, creatinine, phosphate, H⁺, potassium, unmeasured anions, low-molecular-weight proteins, lipids, and carbohydrates.
1. Fluid and Electrolyte Disturbances
| Abnormality | Mechanism | Onset (CKD stage) |
|---|---|---|
| Hypertension | Na/H₂O retention + ↑ systemic vascular resistance (↓ NO) + RAS/SNS activation | G1-G2 (early) |
| Oedema | Na/H₂O retention due to reduced nephron excretory capacity | G3-G5 |
| Hyperkalaemia | ↓ filtered load + ↓ tubular secretion. Compounded by type 4 RTA, ACEi/ARBi, metabolic acidosis | G3b-G4 |
| Hyponatraemia | Free water retention, impaired diluting capacity | G4-G5 |
2. Acid-Base Disorder
- Uremic acidosis (most common): Decreased ammonia production from reduced nephron mass → inability to excrete H⁺ → metabolic acidosis
- Mechanism: ↓ NH₃ synthesis → ↓ urinary buffering → H⁺ accumulation
- Initially normal anion gap (hyperchloraemic)
- At GFR <25 mL/min (Stage G4-G5): elevated anion gap as phosphate, sulfate, and organic anions accumulate
- Serum bicarbonate typically stabilises at 12-18 mEq/L (bone acts as buffer)
- Causes of early-stage acidosis:
- Type 1 RTA (distal: impaired H⁺ secretion, non-acid urine, hypokalemia)
- Type 2 RTA (proximal: impaired HCO₃⁻ reabsorption, hypokalemia)
- Type 4 RTA (hyporeninemic hypoaldosteronism: acid urine, hyperkalemia - most common in diabetic nephropathy)
3. Calcium, Phosphate, and Bone Disease (CKD-MBD)
This is one of the earliest and most important consequences of CKD:
Pathophysiology cascade:
- ↓ GFR → phosphate retention (phosphate cannot be excreted)
- ↑ Serum phosphate → directly stimulates PTH secretion
- ↓ Renal 1α-hydroxylase activity → ↓ 1,25-(OH)₂ Vitamin D (calcitriol)
- ↓ Calcitriol → reduced intestinal Ca²⁺ absorption → hypocalcaemia
- Hypocalcaemia + hyperphosphataemia → further stimulate secondary hyperparathyroidism
- PTH causes bone resorption (osteitis fibrosa cystica), but also vitamin D deficiency causes defective mineralisation (osteomalacia)
- Fibroblast Growth Factor-23 (FGF-23) rises very early in CKD (before PTH rises) as a phosphaturic factor; elevated FGF-23 is independently associated with left ventricular hypertrophy and cardiovascular mortality
- Ca × PO₄ product elevation → vascular and soft tissue calcification (metastatic calcification)
- In ESKD: autonomous PTH secretion = tertiary hyperparathyroidism
Onset: PTH and vitamin D abnormalities begin at stage G3a - earlier than most other complications
4. Anaemia of CKD
- Mechanism: ↓ erythropoietin (EPO) production by peritubular fibroblasts of the kidney (main driver)
- Compounded by: ↓ red cell survival, iron deficiency (absolute or functional - elevated hepcidin blocks iron absorption and release from macrophages), deficiency of folate/B12, blood loss from dialysis
- Normochromic, normocytic (or mildly microcytic if iron-deficient)
- Appears at stage G3a-G3b; becomes significant (Hb <10 g/dL) at stage G4
- Contributes to: fatigue, reduced exercise tolerance, left ventricular hypertrophy (compensatory for tissue hypoxia)
5. Cardiovascular Complications
CKD is an independent risk factor for cardiovascular disease:
| Mechanism | Consequence |
|---|---|
| Fluid/Na retention | Hypertension, LVH, HF |
| RAS/SNS activation | Hypertension, LVH, arrhythmia |
| ↑ FGF-23 | LVH (direct effect on cardiomyocytes) |
| Dyslipidaemia | ↑ triglycerides, ↓ HDL - accelerated atherosclerosis |
| ↑ CRP, IL-6 | Chronic inflammation → endothelial damage |
| Vascular calcification | Arterial stiffness, coronary disease |
| Anaemia | LVH (compensatory), demand ischemia |
| Hyper-homocysteinaemia | Endothelial dysfunction, thrombosis |
| Hypercoagulability | DVT, PE (in nephrotic range proteinuria) |
CKD patients have 5-10x higher cardiovascular mortality than age-matched controls.
6. Uraemic Neurotoxicity
- At GFR <15 mL/min (stage G5): accumulation of uraemic toxins (particularly middle molecules, phenols, indoles, and guanidino compounds) causes:
- Peripheral neuropathy (glove-and-stocking sensorimotor; "restless legs syndrome")
- Uraemic encephalopathy (asterixis, confusion, seizures, coma)
- Autonomic neuropathy
- Elevated prolactin levels (normally cleared by kidney) → sexual and reproductive dysfunction
7. Uraemic Bleeding Tendency
- Platelet dysfunction (qualitative defect): uraemic toxins impair platelet-vWF binding (GPIb-vWF interaction) and platelet degranulation
- Anaemia worsens bleeding (RBCs normally promote platelet margination to vessel wall)
- Results in prolonged bleeding time despite normal platelet count, PT, and aPTT
8. Immunodeficiency
- Uraemia is an immunocompromised state
- Impaired neutrophil and T-cell function → increased susceptibility to infections (especially encapsulated organisms, TB)
- Hypogammaglobulinaemia in nephrotic states
F. CKD Staging and Biochemical Manifestations by Stage
| GFR Stage | eGFR (mL/min/1.73m²) | Key Biochemical Events |
|---|---|---|
| G1 | ≥90 (with damage marker) | Hypertension begins; no biochemical changes yet |
| G2 | 60-89 | Mild rise in PTH; FGF-23 rises early |
| G3a | 45-59 | PTH ↑, Vit D ↓, anaemia starts, metabolic acidosis (RTA types) |
| G3b | 30-44 | Hyperkalemia starts; acidosis worsens; phosphate rising |
| G4 | 15-29 | Hyperphosphataemia, severe anaemia (Hb <10), high AG acidosis |
| G5 (Uraemia) | <15 | Full uraemic syndrome; indication for RRT planning |
G. Summary of Pathophysiological Mechanisms
PRIMARY INSULT (diabetes, hypertension, GN, etc.)
↓
Nephron loss
↓
Surviving nephrons: HYPERFILTRATION (SNGFR ↑, glomerular HTN)
↓
Proteinuria → tubular injury → TGF-β ↑ → tubulointerstitial fibrosis
↓
Progressive nephron loss (self-perpetuating cycle)
↓
↓ GFR: Retention of H⁺, K⁺, PO₄³⁻, Na⁺, H₂O, uraemic toxins
↓
Metabolic acidosis | Hyperkalaemia | CKD-MBD | Anaemia | CVD | Uraemia
Sources: Goldman-Cecil Medicine, 26e (pp. 1173-1180, 1337-1344) | Washington Manual of Medical Therapeutics | Miller's Anesthesia, 10e | Brenner and Rector's The Kidney | NKF Primer on Kidney Diseases, 8e | Rosen's Emergency Medicine
This is a shared conversation. Sign in to Orris to start your own chat.