IgA nephropathy long note
Reading File
Finding Sources
Finding Sources
Searching PubMed
"IgA nephropathy"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have all the information needed to write a comprehensive long note on IgA nephropathy. Let me compile this now.
IgA Nephropathy (IgAN) - Comprehensive Long Note
Definition and Overview
IgA nephropathy (IgAN), also called Berger disease (after Jean Berger who described it in 1968), is the most common primary glomerulonephritis worldwide. It is defined by the mesangial deposition of IgA-dominant immune complexes, predominantly polymeric IgA1, leading to glomerular inflammation and progressive kidney injury. It accounts for >45% of primary glomerular diseases in China and represents the leading cause of recurrent hematuria diagnosed by renal biopsy globally.
- Comprehensive Clinical Nephrology, 7th Edition, p. 331
- NKF Primer on Kidney Diseases, 8e, p. 3054
- Robbins & Kumar Basic Pathology, p. 512
Epidemiology
| Parameter | Details |
|---|---|
| Global prevalence | Most common primary GN worldwide |
| Incidence | 2-10 per 100,000 person-years |
| Asia (Japan) | 45 cases/million population/year |
| Europe (France) | 31 cases/million population/year |
| USA | Less common in Blacks vs. Whites of European origin |
| Sex ratio | Males:Females = 2:1 to 3:1 in Whites; approaches 1:1 in Asians |
| Age of onset | Typically 2nd-4th decade |
The prevalence in biopsied populations is likely an underestimate. A study of Japanese kidney donors found a prevalence of mesangial IgA deposits with proliferative changes in 1.6% of the general population, suggesting many cases spontaneously remit without ever coming to medical attention.
- Comprehensive Clinical Nephrology, 7th Edition, p. 332
Pathogenesis - The "Four-Hit" Model
IgAN pathogenesis is best understood as a multi-step process:
Hit 1: Elevated Galactose-Deficient IgA1 (Gd-IgA1)
- IgA1 carries O-linked sugars at its hinge region. In IgAN, these are enriched in galactose-deficient IgA1 (gd-IgA1) - where N-acetylgalactosamine is exposed instead of being capped by galactose.
- Excess mucosal IgA1 enters the systemic circulation - either directly from the mucosa-associated lymphoid tissue (MALT) or after translocation of mucosal lymphocytes to the bone marrow.
- Gd-IgA1 is elevated in >90% of patients but is also found in unaffected relatives, confirming it is necessary but not sufficient.
Hit 2: Production of Autoantibodies Against Gd-IgA1
- IgA and IgG autoantibodies form against the exposed N-acetylgalactosamine (GalNAc) residues of gd-IgA1, recognizing it as "foreign."
- These are typically cross-reactive against antigens from microorganisms involved in upper respiratory/gastrointestinal infections (explaining the synpharyngitic hematuria).
Hit 3: Formation and Deposition of Immune Complexes
- Circulating IgA1-anti-IgA1 immune complexes form, which then deposit in the mesangium.
- Deposition may also result from altered IgA1 interactions with matrix proteins, mesangial cell/monocyte Fc receptors (FcαRI), and impaired clearance via hepatic asialoglycoprotein receptors and Kupffer cell FcαR.
- Mesangial IgA1 in IgAN is also enriched for gd-IgA1.
Hit 4: Mesangial Cell Activation and Injury
- Deposited immune complexes activate complement (via alternative and lectin pathways - classical pathway components are typically absent).
- Mesangial cells are activated, producing cytokines, chemokines, and profibrotic mediators (TGF-β, PDGF), leading to mesangial proliferation and progressive fibrosis.
Genetic Factors
- Over 25% of blood relatives have elevated gd-IgA1; familial cases exist but >90% are sporadic.
- Genome-wide association studies (GWAS) have identified risk loci including: HLA-DR, -DQ, -DP, HLA-B; α-defensin (DEFA) gene cluster; complement factor H; CARD8; MYCT1; ZNF543.
- Geographic variation mirrors genetic susceptibility: highest risk in East Asia, lower in Europe/North America.
Role of Mucosal Immunity
-
Close association between mucosal infections and episodes of hematuria ("synpharyngitic" hematuria - hematuria concurrent with or within 1-2 days of upper respiratory infection).
-
Altered gut microbiome composition in IgAN.
-
Gut bacteria modulate IgA class switching, mucosal IgA production, and IgA1 O-galactosylation.
-
Risk alleles from GWAS overlap with inflammatory bowel disease susceptibility genes.
-
Comprehensive Clinical Nephrology, 7th Edition, pp. 331-332
-
Robbins & Kumar Basic Pathology, p. 512
-
Brenner and Rector's The Kidney, p. 3378
Clinical Manifestations
IgAN has a wide spectrum of presentations:
1. Episodic Gross (Macroscopic) Hematuria - Most Common Presentation in Young Adults (~50%)
- Coca-cola/tea-colored urine, often concurrent with or within 24-48 hours of an upper respiratory tract infection (synpharyngitic hematuria).
- Contrasts with post-streptococcal GN where hematuria appears 1-3 weeks after infection.
- Episodes typically resolve spontaneously; interval hematuria may persist.
2. Asymptomatic Microscopic Hematuria ± Proteinuria
- More common in older adults or those identified by routine urinalysis screening.
- Persistent microscopic hematuria is the most common finding in population screening.
3. Nephrotic Syndrome (Rare)
- Nephrotic-range proteinuria occurs but is unusual; should prompt investigation for a concurrent podocytopathy (e.g., minimal change disease superimposed on IgAN).
4. Acute Kidney Injury (<5% of all cases)
- Three mechanisms:
- Crescentic (rapidly progressive) IgAN - necrotizing GN with crescent formation.
- Tubule occlusion by red blood cells from heavy glomerular hematuria.
- Incidental AKI superimposed on chronic IgAN (e.g., NSAID-induced AIN) - more common in elderly.
- AKI presentation carries a significantly higher risk of progression to ESKD.
5. Chronic Kidney Disease with Hypertension
- Older patients with longstanding undiagnosed disease; already have hypertension and impaired GFR at first presentation.
- Accelerated hypertension occurs in ~5% of patients.
6. IgA Vasculitis (Henoch-Schönlein Purpura / HSP)
- Systemic form of IgA-mediated disease with classic tetrad: palpable purpura (buttocks/legs), arthralgias, abdominal pain, and nephritis.
- Children have a more favorable renal prognosis than adults.
Secondary IgA Nephropathy
Mesangial IgA deposition without typical IgAN occurs in association with:
- Liver disease (especially alcoholic cirrhosis, hepatitis B, schistosomiasis) - due to impaired IgA clearance by hepatic asialoglycoprotein receptors and Kupffer cells.
- Inflammatory bowel disease (IBD increases risk of ESKD in IgAN).
- Celiac disease (mucosal IgA hyperresponsiveness).
- Ankylosing spondylitis
- Dermatitis herpetiformis
- HIV/AIDS (polyclonal IgA increase)
Renal Pathology
Light Microscopy
Lesions are variable; the glomeruli may show:
- Normal appearance (mesangial IgA found only on IF)
- Mesangial widening and hypercellularity (most common)
- Focal proliferative GN (segmental inflammation in some glomeruli)
- Diffuse mesangial proliferative GN
- Crescentic GN (rare, severe disease)
Immunofluorescence (IF) - Diagnostic Hallmark
- Dominant or codominant mesangial IgA deposition (required for diagnosis)
- Often accompanied by C3, properdin (alternative pathway)
- Smaller amounts of IgG or IgM may be present
- Early classical complement components (C1q, C4) are characteristically absent
Electron Microscopy
- Confirms mesangial electron-dense deposits
- Paramesangial deposits may extend into capillary walls in severe disease
IgA Nephropathy Histology (Robbins & Comprehensive Clinical Nephrology)
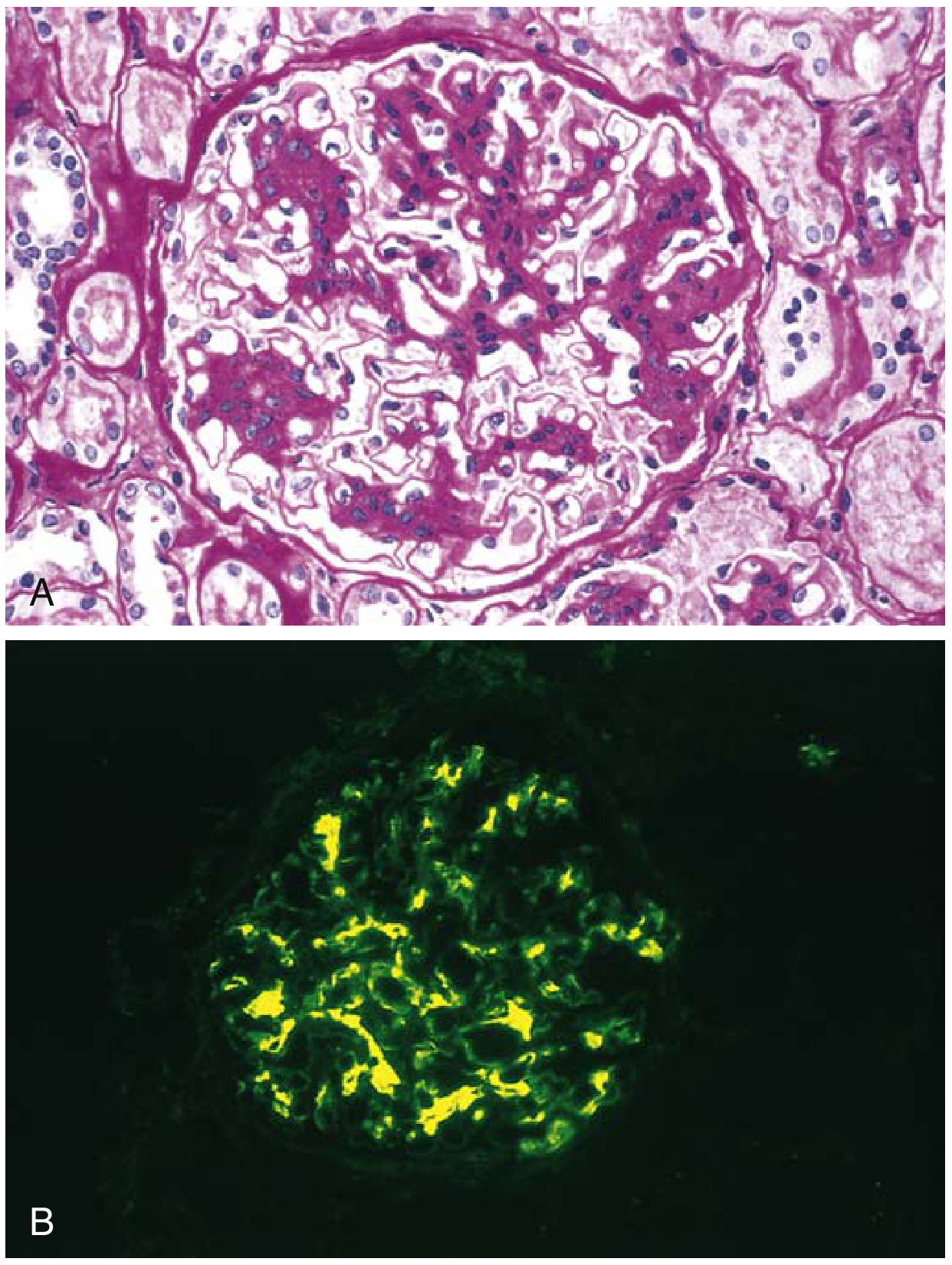
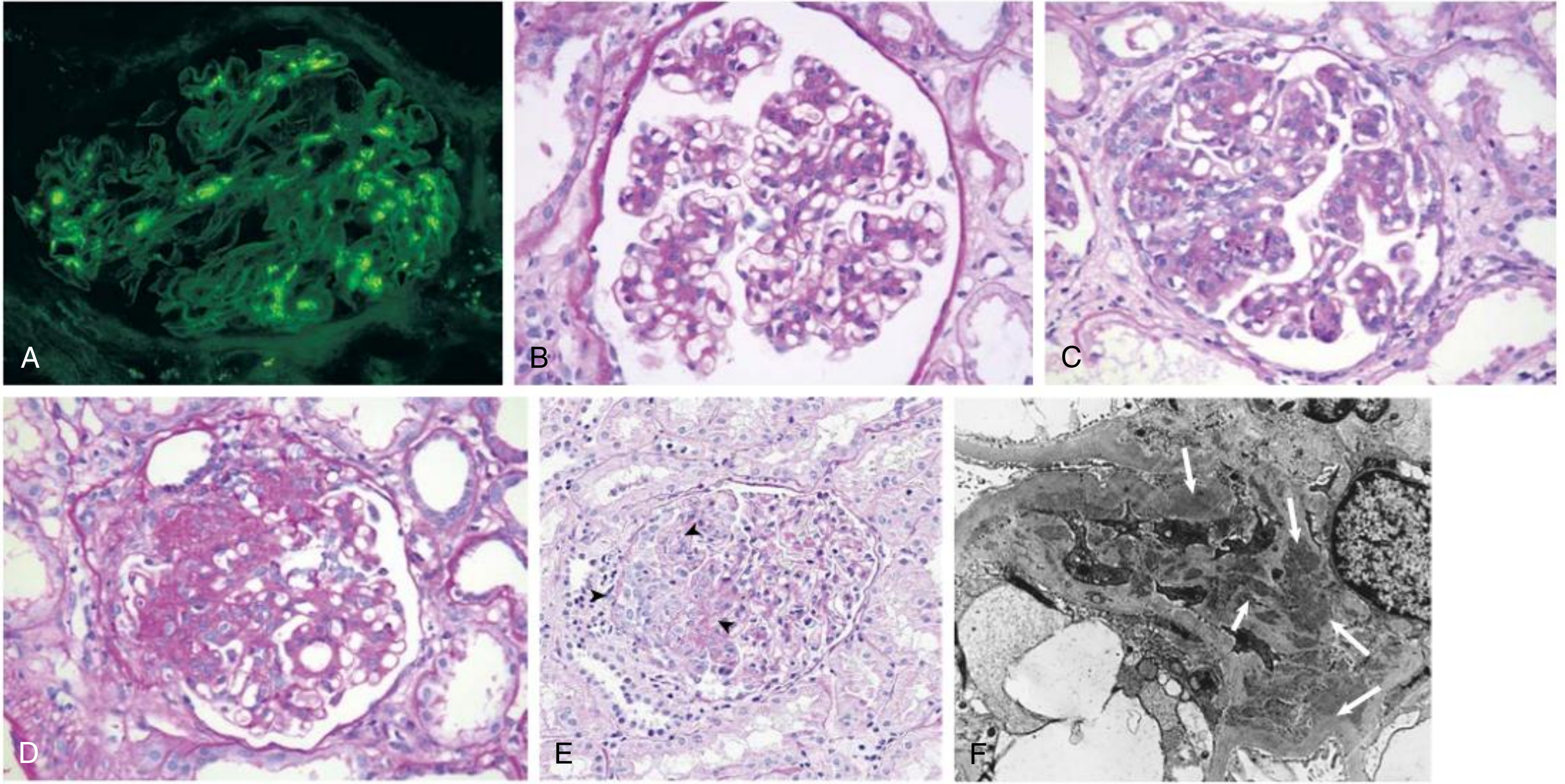
The Oxford Classification (MEST-C Score)
The Oxford classification of IgAN (2009, updated 2016) provides standardized pathologic scoring that carries independent prognostic value beyond clinical parameters:
| Variable | Description | Score | Prognostic Significance |
|---|---|---|---|
| M - Mesangial hypercellularity | >4 mesangial cells in any mesangial area | M0 (<50% glomeruli) / M1 (>50%) | M1 = worse prognosis |
| E - Endocapillary proliferation | Hypercellularity causing luminal occlusion | E0 / E1 | E1 = worse, responsive to steroids |
| S - Segmental sclerosis | Any portion of tuft involved in sclerosis/adhesion | S0 / S1 | S1 = worse prognosis |
| T - Tubular atrophy/interstitial fibrosis | % cortical area | T0 (<25%) / T1 (25-50%) / T2 (>50%) | T score = most consistent predictor of ESKD |
| C - Crescents | Cellular or fibrocellular crescents | C0 / C1 (<25% glomeruli) / C2 (>25%) | C1/C2 = worse, may respond to IS |
T score provides the most consistent guide for progression to ESKD across all studies.
The International IgAN Prediction Tool (www.qxmd.com) combines clinical risk factors (GFR, proteinuria, hypertension) with Oxford MEST scores to predict 50% decline in eGFR or ESKD at selected time intervals.
- Comprehensive Clinical Nephrology, 7th Edition, p. 314
- NKF Primer on Kidney Diseases, 8e, p. 3060
Diagnosis
- No specific serum or urine biomarker exists for IgAN.
- Renal biopsy is required for definitive diagnosis.
- Serum IgA levels are elevated in ~50% of patients but are non-specific and not diagnostic.
- Serum gd-IgA1 levels are elevated in most IgAN patients but also in relatives without nephropathy.
Differential Diagnosis
| Condition | Distinguishing Feature |
|---|---|
| Post-streptococcal GN | Hematuria 1-3 weeks after infection; low C3; resolves |
| Alport syndrome | Family history; GBM thinning/splitting on EM; COL4A mutations |
| Thin GBM disease | Diffuse GBM thinning; no mesangial deposits on IF |
| IgA vasculitis (HSP) | Systemic features (purpura, arthritis, GI); identical renal biopsy |
| Lupus nephritis | ANA/anti-dsDNA positive; C3/C4 low; C1q on IF |
| MPGN | Subendothelial/subepithelial deposits; C3 low |
Prognosis and Risk Stratification
About 30-40% of patients reach ESKD within 20-30 years of initial presentation. However, prognosis is highly variable:
Clinical Predictors of Progression (at Biopsy):
- Reduced GFR at presentation
- Hypertension (persistent)
- Proteinuria >1 g/24 hours (especially time-averaged proteinuria)
- Older age at onset
During Follow-up:
- Only persistent hypertension and time-averaged proteinuria are reliable predictors.
- Risk of progression is low when proteinuria remains <0.2 g/24h with normal BP.
- Persistent microscopic hematuria is associated with worse outcomes; hematuria remission improves outcomes in those with persistent proteinuria.
- Hyperuricemia, smoking, and increased BMI are independent risk factors.
Histologic Predictors:
- T score (tubular atrophy/interstitial fibrosis) - most consistent predictor of ESKD.
- High MEST-C scores add prognostic information beyond clinical features.
Low-Risk Features (Mostly Good 7-10 Year Prognosis):
-
Isolated microhematuria
-
Little or no proteinuria
-
Normal BP and GFR at diagnosis
-
Note: Up to 40% of these patients develop increasing proteinuria; up to 5% lose GFR over this period - necessitating regular follow-up.
-
Comprehensive Clinical Nephrology, 7th Edition, pp. 309-316
Treatment
1. Supportive and Kidney-Protective Measures (All Patients)
- Renin-angiotensin system (RAS) blockade: ACE inhibitor or ARB - first-line therapy to reduce proteinuria and slow progression. Target BP <130/80 mmHg.
- SGLT2 inhibitors (e.g., dapagliflozin): Emerging role in reducing proteinuria and preserving GFR.
- Dietary salt restriction, weight loss, smoking cessation.
- Statin therapy for dyslipidemia.
- Fish oil (omega-3 fatty acids): Modest benefit shown in some trials; used in some guidelines for patients with proteinuria >1 g/day.
2. Tonsillectomy
- Tonsillectomy has been practiced in Japan for patients with frequent tonsillitis-associated flares.
- Evidence in Western populations is limited; not widely recommended.
3. Corticosteroids
- Indications: Patients with persistent proteinuria >1 g/24h despite optimal RAS blockade AND eGFR >30 mL/min/1.73m².
- STOP-IgAN trial: Intensive supportive care ± corticosteroids; corticosteroids reduced proteinuria but did not reduce rate of GFR decline, with significant infections and metabolic side effects.
- TESTING trial (2017 and extended 2022): Methylprednisolone significantly reduced risk of kidney failure compared to placebo in patients with proteinuria >1 g/day (eGFR 20-120), but with significantly increased serious adverse events (infections, deaths) at full dose; low-dose methylprednisolone protocol showed better safety.
- KDIGO 2021 guidelines: Consider steroids only in selected high-risk patients with optimal supportive care first; weigh infection risk.
4. Immunosuppression
- Cyclophosphamide + steroids: For crescentic IgAN (rapidly progressive GN presentation); analogous to treatment of ANCA vasculitis-related crescentic GN.
- Mycophenolate mofetil (MMF): Trials largely negative in Western populations; some benefit in Chinese patients.
- Rituximab: Some case reports in children with severe disease; not established standard.
5. Targeted APRIL/BAFF Pathway Therapies (Novel)
- Sparsentan (dual endothelin-angiotensin receptor antagonist): FDA-approved for IgAN.
- Budesonide (Nefecon/Tarpeyo): Targeted-release formulation delivering steroids to the Peyer's patches in the distal ileum, reducing mucosal IgA production with fewer systemic steroid side effects. FDA-approved for IgAN with proteinuria >1 g/day.
- Iptacopan (complement factor B inhibitor), Atacicept (APRIL/BAFF blocker): In clinical trials.
6. Treatment of Specific Presentations
- Crescentic IgAN (RPGN): Pulse methylprednisolone + cyclophosphamide; consider plasma exchange in severe cases (though evidence limited compared to ANCA-GN).
- AKI from hematuria-induced tubular occlusion: Supportive care; RBC casts in tubules.
- IgA vasculitis (HSP) nephritis: Corticosteroids for systemic features; immunosuppression for severe nephritis (EULAR/PRINTO/PRES guidelines).
Transplantation
-
IgAN patients are ideal transplant candidates (relatively young, few comorbidities).
-
Mesangial IgA deposits recur in up to 60% of transplanted kidneys.
-
Recurrence may occur within days to weeks but is often clinically silent initially.
-
Recurrent IgAN: 30% in living-related grafts vs. 23% in cadaveric grafts.
-
Graft survival is not significantly affected in the first 10 years; thereafter, recurrent disease may accelerate graft loss.
-
Transplanting kidneys with IgA deposits into non-IgAN recipients leads to rapid disappearance of deposits, confirming the disease lies in the recipient's immune system, not the kidney.
-
Crescentic recurrent IgAN is rare but resistant to treatment and causes rapid graft failure.
-
Comprehensive Clinical Nephrology, 7th Edition, pp. 319-322
IgAN vs. HSP (IgA Vasculitis) Nephritis
| Feature | Primary IgAN | IgA Vasculitis Nephritis |
|---|---|---|
| Systemic features | Absent | Purpura, arthritis, GI bleeding |
| Renal biopsy | Identical (mesangial IgA) | Identical (mesangial IgA) |
| Age | Young adults | Children > adults |
| Prognosis | Variable | Favorable in children; worse in adults |
| Pathophysiology | Same gd-IgA1 mechanism | Same |
Summary Flowchart
Patient with hematuria ± proteinuria
↓
Urinalysis + 24h urine protein + serum creatinine/eGFR
↓
Persistent hematuria ± proteinuria > 0.5 g/day OR impaired eGFR
↓
Renal Biopsy
↓
Mesangial IgA on IF → IgA Nephropathy
↓
MEST-C scoring + Clinical risk stratification
↓
Low risk: Supportive (RAS blockade, BP control, lifestyle)
High risk (proteinuria >1g, reduced eGFR, M1/E1/T1-2/C1-2):
→ Optimize RAS blockade → Consider steroids/IS
→ Crescentic: Pulse steroids + cyclophosphamide
Key Points to Remember
- Most common primary GN worldwide; highest incidence in East Asia.
- Pathogenesis: gd-IgA1 → autoantibodies → immune complexes → mesangial deposition → complement activation → injury.
- Classic presentation: synpharyngitic gross hematuria in a young male (vs. post-streptococcal hematuria appearing 1-3 weeks later).
- Diagnosis requires renal biopsy - mesangial IgA dominant on immunofluorescence; no C1q or C4.
- Oxford MEST-C score guides prognosis; T score is the most reliable predictor of ESKD.
- 30-40% reach ESKD in 20-30 years; prognosis driven by hypertension, proteinuria, and eGFR.
- Treatment: RAS blockade for all; steroids for high-risk (proteinuria >1g + eGFR >30); cyclophosphamide for crescentic disease; newer agents include budesonide (Nefecon) and sparsentan.
- Transplant recurrence in up to 60% but rarely affects graft survival before 10 years.
Sources:
- Comprehensive Clinical Nephrology, 7th Edition - Chapters 24 & 80
- NKF Primer on Kidney Diseases, 8th Edition - Chapter 21
- Robbins & Kumar Basic Pathology - Chapter 12
- Brenner and Rector's The Kidney, 2-Volume Set
This is a shared conversation. Sign in to Orris to start your own chat.