Lesh nyhan syndrome
Reading File
Finding Sources
Searching PubMed
Lesch-Nyhan syndrome HGPRT management
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Here is a comprehensive overview of Lesch-Nyhan Syndrome (LNS).
Lesch-Nyhan Syndrome
Background
Lesch-Nyhan syndrome is a rare X-linked recessive metabolic disorder. It was described by Lesch and Nyhan in 1964, although the association of hyperuricemia with spasticity and choreoathetosis in early childhood had been noted earlier. It is also known as juvenile gout.
- Adams and Victor's Principles of Neurology, 12th Ed, p. 991
- Andrews' Diseases of the Skin, p. 630
Genetics
| Feature | Detail |
|---|---|
| Inheritance | X-linked recessive |
| Gene | HPRT1 (hypoxanthine phosphoribosyltransferase 1) |
| Chromosomal locus | Xq26-q27 |
| Affected sex | Almost exclusively males; females are carriers |
Mutations causing HPRT deficiency include deletions, frameshift mutations, base substitutions, and aberrant mRNA splicing.
- Adams and Victor's Principles of Neurology, p. 991
Biochemical Mechanism
HPRT (hypoxanthine-guanine phosphoribosyltransferase) is a key enzyme in the purine salvage pathway. It normally recycles hypoxanthine and guanine back to IMP and GMP, respectively.
When HPRT is absent or deficient:
- Hypoxanthine is not salvaged - it is catabolized to xanthine and then uric acid (via xanthine oxidase)
- Intracellular PRPP (phosphoribosyl pyrophosphate) accumulates, driving increased de novo purine synthesis
- This results in massive overproduction of uric acid (hyperuricemia)
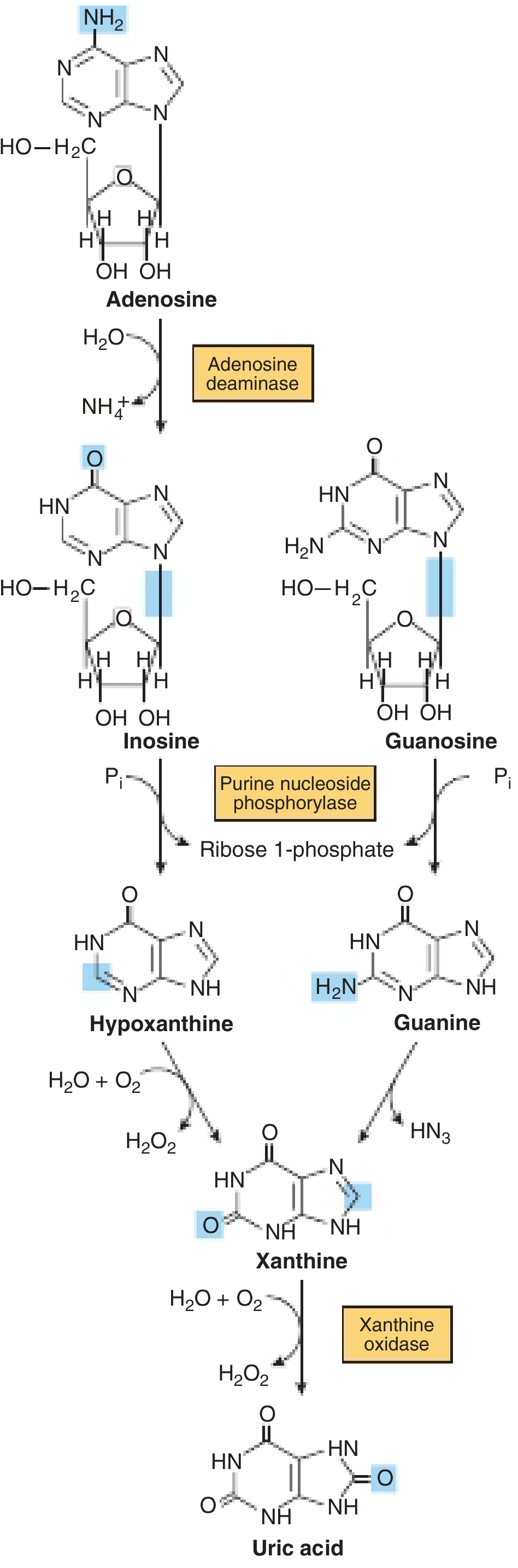
Pathway showing formation of uric acid from purine nucleosides through hypoxanthine and xanthine - Harper's Illustrated Biochemistry, 32nd Ed
- Harper's Illustrated Biochemistry, 32nd Ed, p. 356
Clinical Features
Timeline of Presentation
| Age | Feature |
|---|---|
| Birth to 3-6 months | Appears normal, develops on schedule |
| 3-6 months | Maturational delay begins; hypotonia |
| Later infancy | Hypotonia gives way to hypertonia |
| 2nd-3rd year | Compulsive self-mutilation (biting lips, fingers) |
| Later childhood | Spasticity, choreoathetosis, tremor |
| >10 years | Gouty tophi on ears; risk of gouty nephropathy |
Key Features (Classic Triad)
- Neurological dysfunction: choreoathetosis, spasticity, dystonia, dysarthric speech, moderate-to-severe intellectual disability
- Behavioral abnormality: compulsive self-mutilation (lips and fingers bitten; ears and nose occasionally mutilated), aggression
- Hyperuricemia and gout: uric acid levels 7-10 mg/dL; uric acid lithiasis; gouty nephropathy
Early Diagnostic Clue
- Orange urate crystals in the diaper - a hallmark early finding
Skin/Tophi
- Gouty tophi visible on the ear (shown below)
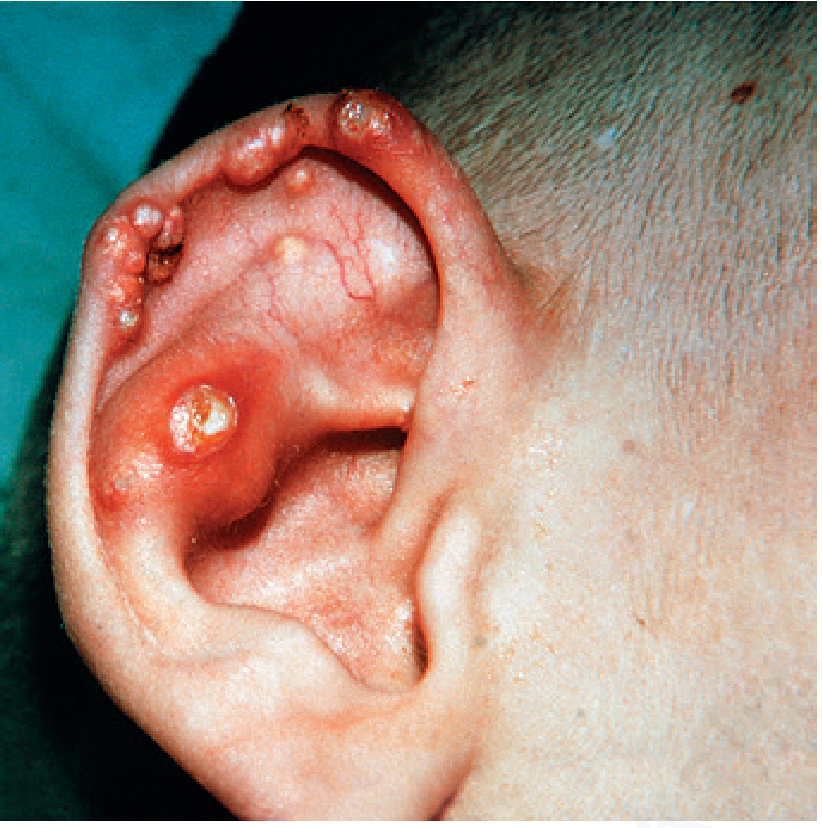
Fig. 26.40 - Lesch-Nyhan syndrome showing gouty tophi on the ear - Andrews' Diseases of the Skin
Pathophysiology of CNS Dysfunction
The exact mechanism by which HPRT deficiency causes neurological and behavioral abnormalities is unclear. However:
- Both GMP and IMP are deficient in LNS
- Dopaminergic pathways are implicated (LNS appears in differential diagnosis of acquired dystonia related to dopamine metabolism disorders)
- The self-mutilation behavior is compulsive, not pain-insensitivity-based - patients are distressed by their own actions
Diagnosis
- Serum uric acid: 7-10 mg/dL (elevated)
- HPRT enzyme assay (in red blood cells): markedly reduced or absent activity
- Genetic analysis of the HPRT1 gene: confirms diagnosis; detects female carriers
- Early clue: orange/sandy urate crystals in diaper/urine
Differential Diagnosis:
- Non-specific intellectual disability/autism with self-injurious behavior
- Athetosis from birth trauma
- Encephalopathies with chronic renal disease
- Other purine/pyrimidine metabolism disorders with hyperuricemia
Treatment
There is no cure. Management is largely symptomatic:
| Target | Agent | Notes |
|---|---|---|
| Hyperuricemia / gout / nephropathy | Allopurinol (200-400 mg/day) | Xanthine oxidase inhibitor; prevents uricosuric nephropathy; does NOT help CNS symptoms |
| Self-mutilation | Fluphenazine (Prolixin) | Reported to suppress self-mutilation when haloperidol failed |
| Behavioral control | Haloperidol, behavior modification | Limited benefit |
| Dystonia | Symptomatic treatment | Standard dystonia agents |
| Attempted (no benefit) | GMP + IMP replacement | Replacement of deficient purines did not help |
| Transitory success | 5-hydroxytryptophan + L-dopa | Limited/transient effect |
- Physical restraints are often used to prevent self-inflicted injury
- Adams and Victor's Principles of Neurology, p. 991
Recent Evidence
A 2024 systematic review (PMID: 39117131) specifically assessed management of neurological symptoms in Lesch-Nyhan disease, confirming that controlling hyperuricemia does not address the neurological burden, and highlighting the ongoing challenge of treating the CNS manifestations.
Summary Table
| Feature | Detail |
|---|---|
| Inheritance | X-linked recessive |
| Deficient enzyme | HPRT (hypoxanthine-guanine phosphoribosyltransferase) |
| Gene/locus | HPRT1 / Xq26-q27 |
| Core biochemical defect | Failed purine salvage → uric acid overproduction + PRPP accumulation |
| Key labs | Uric acid 7-10 mg/dL, absent HPRT activity |
| Hallmark behavior | Compulsive self-mutilation |
| Hallmark early sign | Orange urate crystals in diaper |
| First-line for hyperuricemia | Allopurinol |
| CNS treatment | No effective treatment; supportive only |
This is a shared conversation. Sign in to Orris to start your own chat.