Complement system in immunity
Reading File
Finding Sources
Searching PubMed
complement system immunity activation pathways
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Finding Sources
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks and excellent diagrams. Let me compile the full answer.
The Complement System in Immunity
Overview
The complement system is a collection of more than 20 plasma proteins (numbered C1-C9, plus regulatory factors) that serve as a major effector arm of both innate and adaptive immunity. The word "complement" comes from Jules Bordet's 19th-century observation that serum contains a heat-labile component that "complements" antibodies in lysing bacteria - antibodies alone could agglutinate bacteria, but fresh serum was needed for lysis.
The proteins are normally present as inactive zymogens (inactive enzyme precursors). Activation triggers sequential proteolytic cascades with massive amplification - millions of C3b molecules can deposit on a microbe within 2-3 minutes. Critically, complement activation is confined to microbial surfaces and sites of antibody-antigen binding; free activation in blood is suppressed by regulatory proteins.
- Cellular and Molecular Immunology, p. 838-839
- Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 97
Three Activation Pathways
All three pathways converge on C3 cleavage, but differ in how they are initiated:
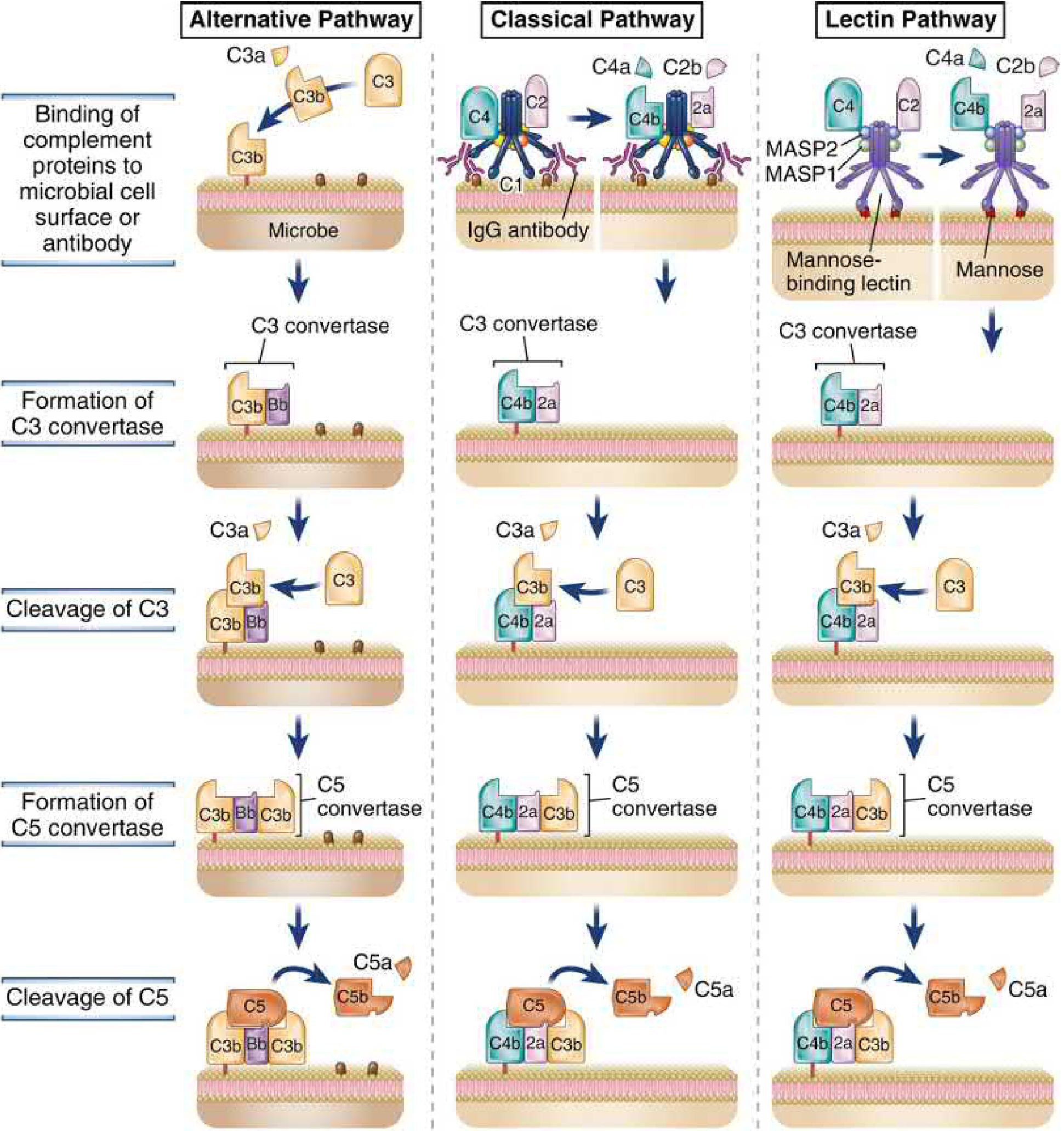
1. Alternative Pathway (Phylogenetically oldest - innate immunity)
- Trigger: C3 spontaneously hydrolyzes in plasma at low levels ("tick-over"), and C3b binds to any nearby surface. On host cells, regulatory proteins (Factor H, DAF) rapidly degrade C3b. On microbial surfaces (LPS, fungal cell walls, complex polysaccharides), these regulators are absent, so C3b is stabilized.
- C3b binds Factor B, which is cleaved by Factor D to generate the alternative pathway C3 convertase: C3bBb.
- A positive feedback loop amplifies activation - more C3b deposits, generating more C3 convertase.
- This pathway distinguishes self from non-self by the presence/absence of host regulatory proteins.
2. Classical Pathway (Adaptive immunity effector; discovered first)
- Trigger: C1q (part of the C1 complex with C1r and C1s) binds to the Fc region of IgM or IgG already bound to antigen on microbial surfaces. Pentameric IgM is particularly potent (one molecule can fix C1). IgG requires two adjacent molecules.
- Pentraxins (e.g., C-reactive protein, CRP) can also bind C1q and activate this pathway.
- C1r and C1s become activated, then cleave C4 (→ C4a + C4b) and C2 (→ C2a + C2b) to form C3 convertase: C4b2a.
- This is the major complement mechanism linking humoral adaptive immunity to complement effector functions.
3. Lectin Pathway (Innate immunity)
-
Trigger: Mannose-Binding Lectin (MBL), a collectin with structural similarity to C1q, recognizes terminal mannose residues on microbial glycoproteins/glycolipids. (Mammalian proteins are typically sialylated and lack free mannose termini.)
-
MBL associates with MASP1 and MASP2 (functional analogs of C1r/C1s), which activate C4 and C2 in the same manner as the classical pathway.
-
C3 convertase formed: C4b2a (identical to classical pathway).
-
Cellular and Molecular Immunology, p. 840-842
-
Robbins Pathologic Basis of Disease, p. 97-98
The Central Event: C3 Cleavage
All three pathways generate a C3 convertase, which cleaves C3 into:
| Fragment | Fate | Function |
|---|---|---|
| C3a | Released into fluid phase | Anaphylatoxin - mast cell degranulation, histamine release, increased vascular permeability, vasodilation |
| C3b | Covalently binds to microbial surface (thioester bond) | Opsonin - promotes phagocytosis via CR1/CR3 on neutrophils/macrophages |
C3b then combines with the C3 convertase to form the C5 convertase, which cleaves C5:
| Fragment | Fate | Function |
|---|---|---|
| C5a | Released | Potent anaphylatoxin + chemotaxin for neutrophils, monocytes, eosinophils; activates lipoxygenase pathway |
| C5b | Remains on surface | Nucleates membrane attack complex (MAC) assembly |
- Robbins Pathologic Basis of Disease, p. 98
Three Main Effector Functions
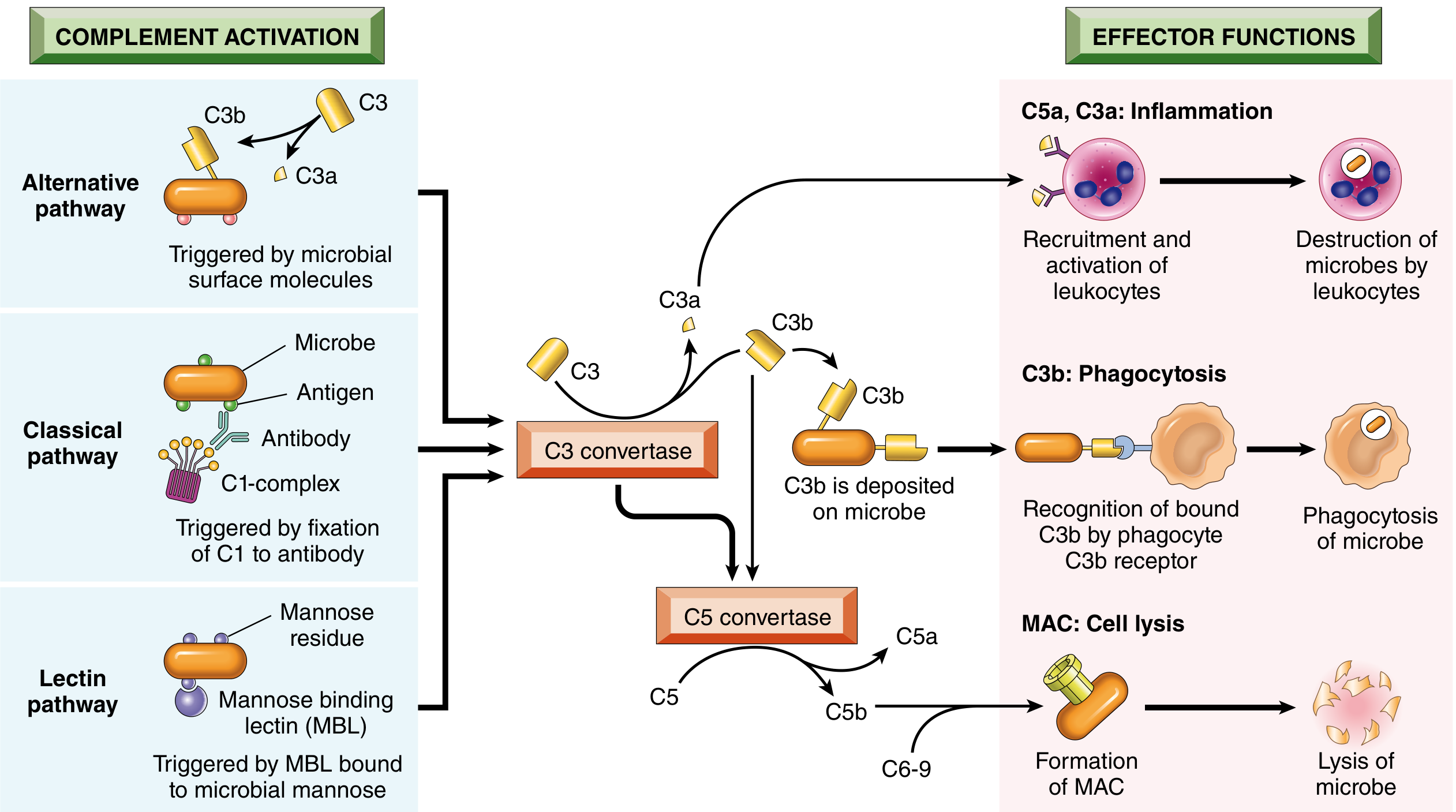
1. Inflammation (C3a, C4a, C5a - "Anaphylatoxins")
- Stimulate histamine release from mast cells and basophils
- Increase vascular permeability and vasodilation
- C5a is the most potent chemotactic factor for neutrophils, monocytes, eosinophils, and basophils
- C5a activates the lipoxygenase pathway in neutrophils/monocytes, amplifying inflammatory mediator release
2. Opsonization and Phagocytosis (C3b, iC3b)
- C3b and its breakdown product iC3b coat microbial surfaces (opsonization)
- Phagocytes express complement receptors: CR1 (recognizes C3b/C4b), CR3/CR4 (recognize iC3b)
- Binding dramatically enhances phagocytic uptake and killing
3. Cell Lysis - Membrane Attack Complex (MAC)
-
C5b recruits C6, C7, C8, and multiple copies of C9 to form the MAC (C5b-9)
-
C9 polymerizes (~10-16 molecules) to form a transmembrane pore (~10 nm diameter)
-
The pore allows free passage of water and ions, causing osmotic lysis
-
Particularly important against gram-negative bacteria with thin cell walls (e.g., Neisseria)
-
Deficiency of terminal complement components (C5-C9) specifically predisposes to disseminated Neisseria infections
-
Robbins Pathologic Basis of Disease, p. 98
Regulation of Complement
Complement regulation is essential to prevent damage to host tissues. Regulatory proteins are expressed on host cells but not on microbes - this is how the system discriminates self from foreign.
| Regulator | Target | Mechanism | Clinical Relevance |
|---|---|---|---|
| C1 inhibitor (C1-INH) | C1r, C1s (classical) | Blocks C1 activation | Deficiency → Hereditary Angioedema (HAE) |
| DAF (CD55) | C3/C5 convertases | Accelerates decay; prevents assembly | GPI-anchored on RBCs/WBCs |
| CD59 | C5b-9 (MAC) | Blocks polymerization of C9 | GPI-anchored on host cells |
| Factor H | Alternative pathway C3b | Promotes cleavage of C3b; turns over C3 convertase | Deficiency/mutations → atypical HUS, age-related macular degeneration |
| C4b-binding protein (C4BP) | C4b (classical/lectin) | Accelerates decay of C4b2a | - |
| MCP (CD46) | C3b, C4b | Cofactor for Factor I-mediated cleavage | Ubiquitous on nucleated cells |
| Factor I | C3b, C4b | Serine protease; cleaves C3b → iC3b (inactive) | Requires cofactor (Factor H or MCP) |
Paroxysmal Nocturnal Hemoglobinuria (PNH): Acquired deficiency of the enzyme PIG-A (which synthesizes GPI anchors) leads to loss of CD55 and CD59 from blood cell surfaces. Erythrocytes become hypersensitive to complement-mediated lysis, causing hemolytic anemia.
- Robbins Pathologic Basis of Disease, p. 98-99
Complement Deficiency States
| Deficiency | Consequence |
|---|---|
| C1q, C1r/s, C4, C2 (early classical) | Susceptibility to encapsulated bacteria; SLE-like autoimmune disease (impaired immune complex clearance) |
| C3 | Most severe - susceptibility to all pyogenic bacteria; recurrent infections |
| Factor D, Factor B, Properdin (alternative) | Susceptibility to Neisseria, encapsulated bacteria |
| C5-C9 (MAC components) | Specific susceptibility to disseminated Neisseria (meningitidis, gonorrhoeae) |
| C1-INH | Hereditary Angioedema - episodic non-pitting bradykinin-mediated edema |
| Factor H / Factor I | Atypical HUS, C3 glomerulopathy, age-related macular degeneration |
Role in Adaptive Immunity
Beyond innate immunity, complement bridges to adaptive immune responses:
- Enhancing B cell activation: C3d (a breakdown product of C3b) on antigen binds CR2 (CD21) on B cells. CR2 is part of the B cell co-receptor complex (CR2-CD19-CD81), which lowers the B cell activation threshold by ~1000-fold. This is why complement dramatically amplifies antibody responses.
- Classical pathway as adaptive effector: IgG and IgM antibodies produced by adaptive immunity activate complement, linking humoral immunity to complement-mediated opsonization and lysis.
- Complement in memory: C3d-coated antigens are trapped by follicular dendritic cells (which express CR1 and CR2), creating antigen depots that sustain long-term B cell memory.
- Immune complex clearance: CR1 on erythrocytes mediates transport of complement-coated immune complexes to the liver and spleen for disposal. Deficiency (as in early-component complement deficiencies) leads to immune complex disease resembling SLE.
Microbial Evasion of Complement
Many pathogens have evolved mechanisms to evade complement:
- Capsules (pneumococcus, meningococcus) physically block C3b deposition
- Sialylation of surface proteins mimics host cells to recruit Factor H
- Staphylococcus aureus secretes SCIN (Staphylococcal Complement Inhibitor) that blocks C3 convertases
- Streptococcus pyogenes expresses M protein, which binds Factor H
- Borrelia burgdorferi outer surface proteins bind complement regulators
- Some viruses encode homologs of host complement regulatory proteins
Summary Table: Three Pathways at a Glance
| Feature | Classical | Lectin | Alternative |
|---|---|---|---|
| Trigger | IgM/IgG-antigen complexes | Mannose on microbes | Microbial surfaces (LPS, fungal walls) |
| Initiator | C1q-C1r-C1s | MBL-MASP1-MASP2 | Spontaneous C3 hydrolysis |
| C3 convertase | C4b2a | C4b2a | C3bBb |
| Immunity type | Adaptive (+ innate) | Innate | Innate |
| Antibody required | Yes | No | No |
| Phylogenetic age | Youngest | Intermediate | Oldest |
- Cellular and Molecular Immunology (Abbas, Lichtman, Pillai), pp. 254-255, 838-842
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 97-99
in more detail
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Here is the detailed, in-depth account of the complement system drawn directly from Cellular and Molecular Immunology (Abbas, Lichtman & Pillai), Robbins Pathologic Basis of Disease, Janeway's Immunobiology, and Roitt's Essential Immunology.
The Complement System in Immunity - Detailed Account
1. Historical Background and Definition
The name "complement" derives from experiments by Jules Bordet (1890s): fresh antibody-containing serum lysed bacteria at 37°C, but heating to 56°C destroyed the lytic activity without affecting agglutination. Adding fresh (non-immunized) serum restored lysis - indicating a heat-labile, non-specific component that "complements" antibody function. This component was later discovered to be a system of more than 20 plasma proteins.
The complement system consists of serum and cell surface proteins that interact in a highly regulated manner to generate products that eliminate microbes. All complement proteins are normally inactive zymogens. Activation proceeds by sequential proteolytic cascades with enormous amplification - each active enzyme at one step generates multiple activated enzyme molecules at the next.
- Cellular and Molecular Immunology, pp. 838-839
2. General Principles of Complement Activation
Four key principles govern complement function:
- Microbe-surface focus: Activation is triggered by microbes and by antibodies/lectins bound to microbes. Complement attacks are therefore directed specifically at microbial surfaces, not in the plasma.
- Proteolytic cascade amplification: Sequential zymogen activation (like the coagulation cascade) produces rapid, massive amplification. Millions of C3b molecules deposit within 2-3 minutes.
- Covalent attachment to targets: Biologically active cleavage products (especially C3b) become covalently attached to microbial surfaces via a reactive thioester bond. In the fluid phase, these bonds hydrolyze within seconds rendering proteins inactive - so full activation only occurs on surfaces.
- Self vs. non-self discrimination: Regulatory proteins on host cells (DAF, CD59, MCP, Factor H) rapidly inactivate any complement components that land on self-cells. Microbes lack these proteins - so complement activation amplifies on microbes but is extinguished on host cells.
3. The Three Activation Pathways
All three pathways converge on the central event: C3 cleavage by a C3 convertase.
3A. The Alternative Pathway (Oldest evolutionarily - innate immunity)
Initiation - C3 Tickover:
C3 (serum concentration 1400-1700 µg/mL) contains a buried reactive thioester bond (Cys-Gln) in its thioester domain. Water spontaneously hydrolyzes this bond at a low rate (1-2% of total plasma C3 per hour) - a process called C3 tickover. This generates C3* (hydrolyzed C3), which binds Factor B. Factor D (a constitutively active serine protease, 1-3 µg/mL) cleaves Factor B within this complex, releasing Ba and leaving Bb attached. This C3*-Bb complex is the fluid-phase C3 convertase.
Amplification on microbial surfaces:
The fluid-phase C3 convertase cleaves C3 to generate C3b. C3b undergoes a dramatic conformational change (~85 Å shift) that exposes the reactive thioester bond. C3b rapidly forms covalent ester or amide bonds with amino/hydroxyl groups on nearby cell surfaces. On host cells, regulatory proteins rapidly inactivate this C3b. On microbial surfaces (which lack regulatory proteins), C3b is stabilized.
Surface-bound C3b binds Factor B, Factor D then cleaves it to generate the surface-bound C3 convertase: C3bBb. Properdin (Factor P) - a dimeric/tetrameric protein (56 kDa subunits) - stabilizes this C3 convertase by binding to C3bBb and significantly prolonging its half-life. The surface-bound C3 convertase cleaves more C3 → generating more C3b → depositing more C3b on the surface → forming more C3 convertase. This positive feedback loop drives massive amplification.
C5 convertase formation (alternative):
When enough C3b accumulates, an additional C3b molecule is recruited to the C3bBb complex, forming the C5 convertase: C3bBbC3b.
Additional facts:
- Sialic acid on mammalian cell surfaces selectively favors binding of Factor H (regulatory) over Factor B (activating) - another self/non-self discrimination mechanism.
- LPS, fungal cell walls, cobra venom factor, complex polysaccharides, and IgA aggregates can trigger this pathway.
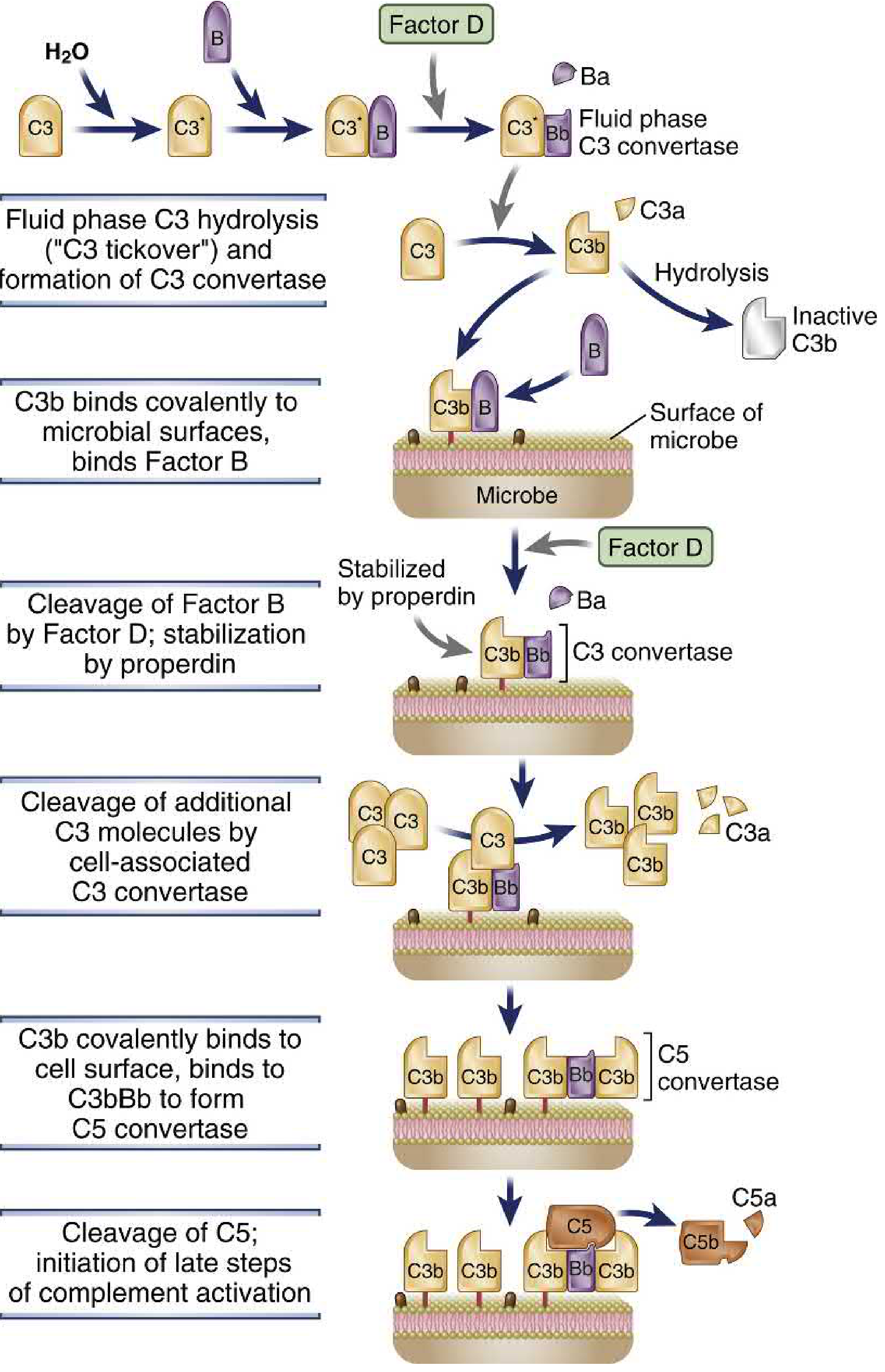
Cellular and Molecular Immunology, pp. 843-845
3B. The Classical Pathway (Adaptive immunity effector - discovered first)
Initiation:
C1 is a macromolecular complex composed of:
- C1q: 6 globular heads connected by collagen-like stalks, arranged like a bunch of tulips. Each head can bind to the Fc region (CH2 domain) of IgG or the Fc region of IgM.
- C1r: two molecules forming a tetramer with C1s
- C1s: two molecules; the effector serine protease
For activation, C1q requires binding of at least two of its globular heads to Ig Fc regions simultaneously. This geometry explains why:
- IgM (pentameric): extremely efficient because a single molecule provides 5 potential Fc sites in close proximity → 1 IgM molecule suffices
- IgG (monomeric): requires two adjacent IgG molecules bound to antigen → less efficient
Among IgG subclasses: IgG3 > IgG1 > IgG2; IgG4 does not fix complement.
C1q binding induces conformational changes in C1r, which autoactivates and then cleaves and activates C1s. Active C1s cleaves:
- C4 → C4a (released, weak anaphylatoxin) + C4b (covalently binds to nearby surface via thioester bond)
- C2 → C2b (released) + C2a (remains bound to C4b)
This forms the Classical pathway C3 convertase: C4b2a
C1 Inhibitor (C1-INH) is a serine protease inhibitor (serpin) that mimics the natural substrate of C1r/C1s. It becomes a "suicide substrate" - cleaved by and covalently linked to C1r and C1s, causing the C1r-C1s tetramer to dissociate from C1q and stopping activation.
C5 convertase (classical): C4b2aC3b
Also: Pentraxins (CRP, serum amyloid P) can bind C1q and activate the classical pathway, linking acute-phase responses to complement.
Cellular and Molecular Immunology, pp. 845-847
3C. The Lectin Pathway (Innate immunity)
Initiation:
Mannose-binding lectin (MBL) is a member of the collectin family with a hexameric structure similar to C1q. MBL recognizes and binds terminal mannose and N-acetylglucosamine residues on microbial glycoproteins and glycolipids. Human cells do not display such sugar patterns (they are typically capped with sialic acid or galactose).
On binding to microbial surfaces, MBL associates with:
- MASP1 (MBL-associated serine protease 1) - functional analog of C1r
- MASP2 (MBL-associated serine protease 2) - functional analog of C1s
MASP2 cleaves C4 and C2 in exactly the same manner as C1s, generating the identical C3 convertase: C4b2a.
Other lectins: Ficolins (L-ficolin, H-ficolin, M-ficolin) recognize acetylated sugars (N-acetylglucosamine, N-acetylgalactosamine) and can also activate the lectin pathway via MASP2.
The subsequent steps are identical to the classical pathway.
4. The Common Terminal Pathway - C5 Cleavage to MAC
C5 Convertase (C3bBbC3b for alternative; C4b2aC3b for classical/lectin):
C5 (190 kDa heterodimer) is cleaved to:
- C5a (small, released): most potent anaphylatoxin - ~20-fold more potent than C3a; chemotaxin for neutrophils/monocytes/eosinophils/basophils; acts directly on endothelium
- C5b (large, surface-bound): initiates MAC assembly
MAC Assembly (C5b → C5b-9):
| Step | Event |
|---|---|
| C5b | Loosely associates with the C3b on the surface |
| + C6 | C6 binds C5b; stabilizes it |
| + C7 | C7 joins; the C5b-7 complex undergoes conformational change exposing hydrophobic domains; inserts into the lipid bilayer |
| + C8 | C8 (trimer: α, β, γ chains) joins; C8α inserts into the membrane, forming a small, unstable pore |
| + C9 (×10-16) | C8 induces conformational change in C9 from hydrophilic to amphipathic; C9 inserts and polymerizes into an annular cylinder (poly-C9) |
The completed MAC (C5b-C6-C7-C8-poly-C9) forms a transmembrane pore ~10 nm in diameter, fully permeable to water and electrolytes. Due to the high intracellular colloid osmotic pressure, there is net influx of Na⁺ and water → osmotic lysis.
Below: MAC formation step-by-step (Cellular and Molecular Immunology) and electron micrograph (Roitt's) showing the annular pore structure
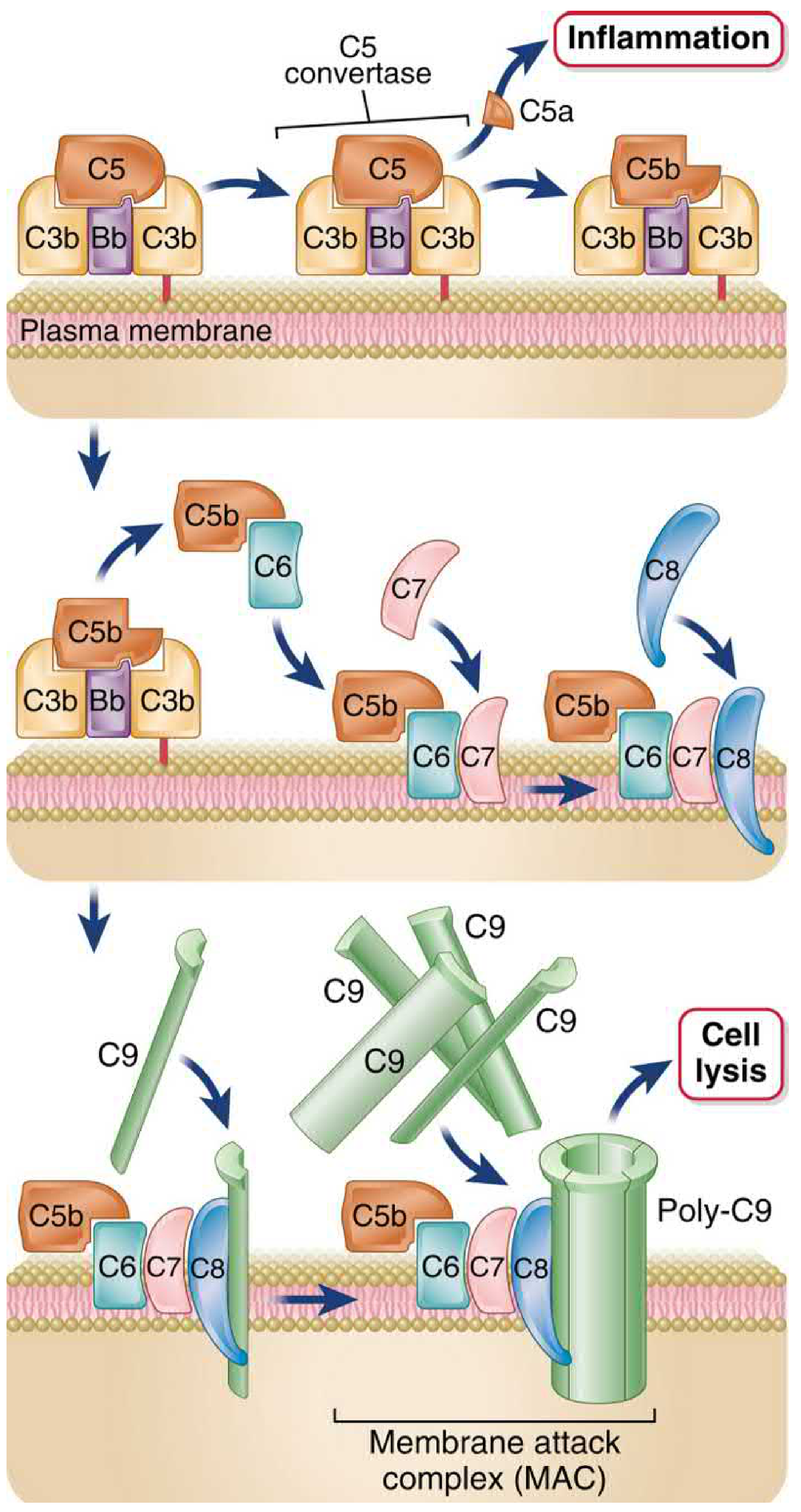
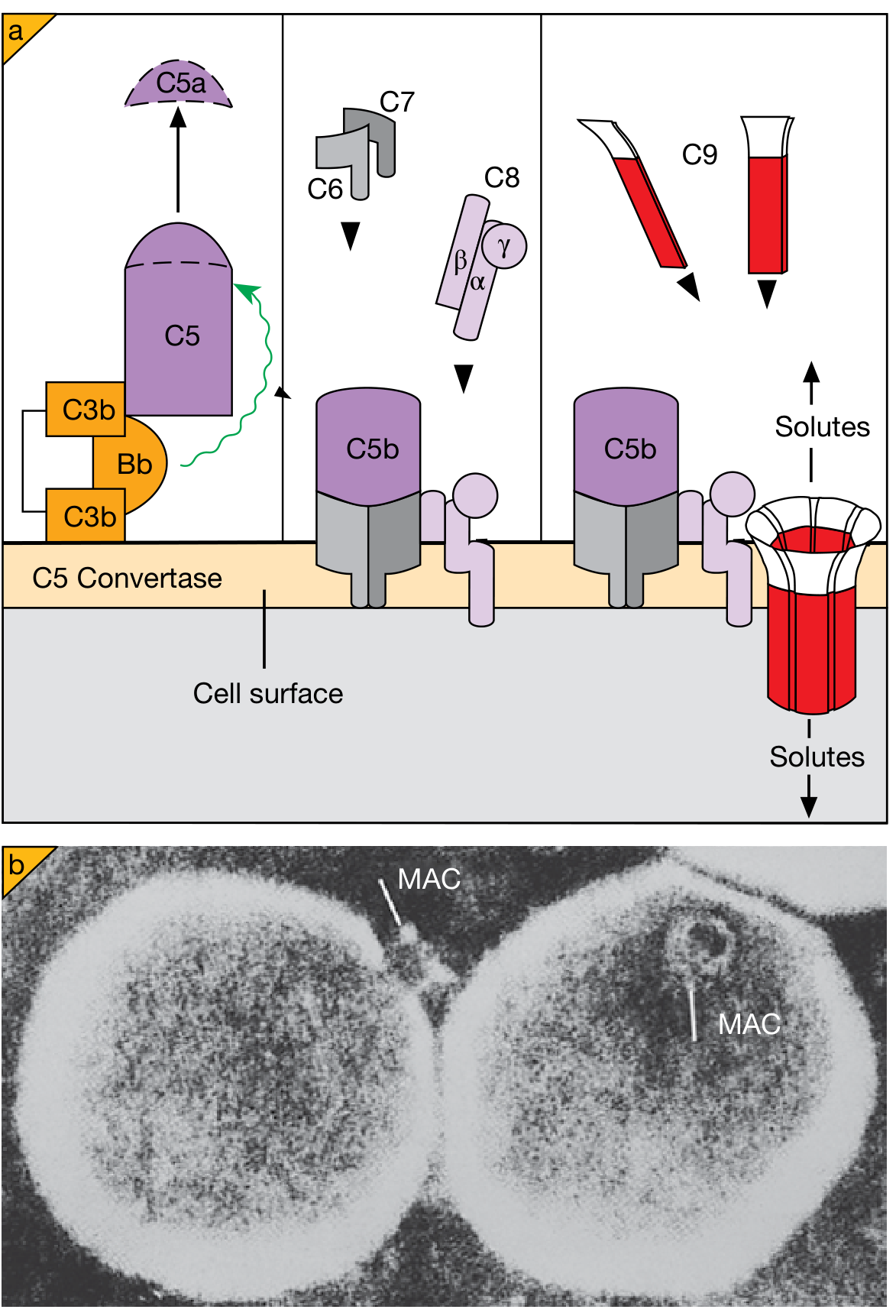
Important nuances about lysis:
- Most bacteria have evolved thick cell walls or capsules preventing MAC access to their membranes
- MAC-mediated killing is critically important only for Neisseria species (very thin cell walls): deficiency of C5-C9 gives specific susceptibility to N. meningitidis and N. gonorrhoeae
- C9 polymerization is structurally homologous to perforin used by cytotoxic T cells, indicating evolutionary conservation of this lytic mechanism
Cellular and Molecular Immunology, p. 857-858; Roitt's Essential Immunology, p. 850-860
5. Complement Receptors - Detailed
The effector functions of complement depend on receptors for complement fragments expressed on various cells:
| Receptor | Ligand | Distribution | Functions |
|---|---|---|---|
| CR1 (CD35) | C3b, C4b, iC3b | Erythrocytes, neutrophils, monocytes/macrophages, B cells, FDCs | (1) Promotes phagocytosis (requires co-stimulus - IFN-γ or C5a); (2) Negative regulator - cofactor for Factor I cleavage of C3b→iC3b; (3) On erythrocytes: transports immune complexes to liver/spleen for clearance |
| CR2 (CD21) | C3d, iC3b, C3dg | B lymphocytes, FDCs | (1) Part of B cell co-receptor complex (CR2-CD19-CD81) - amplifies BCR signaling by ~1000-fold; (2) EBV receptor (EBV uses CR2 to infect B cells) |
| CR3 (Mac-1, CD11b:CD18) | iC3b | Macrophages, monocytes, neutrophils, FDCs | Stimulates phagocytosis; IMPORTANT: CR3 is a β2-integrin - also mediates cell adhesion to endothelium (ICAM-1) |
| CR4 (CD11c:CD18) | iC3b | Macrophages, monocytes, neutrophils, DCs | Stimulates phagocytosis |
| CRIg | C3b, iC3b | Tissue-resident and hepatic sinusoid macrophages (Kupffer cells) | Phagocytosis of circulating pathogens; key in liver complement clearance |
| C5aR1 (CD88) | C5a | Neutrophils, macrophages, endothelial cells, mast cells | 7-transmembrane GPCR; activates neutrophil motility, respiratory burst; mast cell degranulation; endothelial P-selectin upregulation |
| C5aR2 | C5a | Neutrophils, macrophages | Decoy receptor - negative regulator of C5a signaling |
| C3aR | C3a | Macrophages, endothelial cells, mast cells | GPCR; mast cell degranulation; eosinophil chemotaxis |
Synergy between CR1 and C5aR1: CR1 alone is inefficient at triggering phagocytosis. But when macrophages are activated by C5a (binding C5aR1), they engulf bacteria that are simultaneously bound via CR1. This is a critical synergy mechanism.
Cellular and Molecular Immunology, pp. 858-862; Janeway's Immunobiology, pp. 1462-1469
6. Regulatory Proteins - Detailed Mechanisms
Regulation occurs at three levels: (a) early - C1 activation, (b) middle - C3/C5 convertase assembly, (c) late - MAC formation
6a. Inhibition of C1 Activation
C1 Inhibitor (C1-INH): A serpin (serine protease inhibitor). It mimics C1r/C1s substrates and becomes covalently attached to active C1r and C1s → the entire C1r₂-C1s₂ tetramer dissociates from C1q. This limits: (a) classical pathway and (b) lectin pathway (via MASP2 inhibition). C1-INH also inhibits kallikrein and Factor XII, limiting bradykinin production.
Hereditary Angioedema (HAE): Autosomal dominant C1-INH deficiency. Uncontrolled C1 activation → C2/C4 consumption → generation of C2 kinin and bradykinin → episodic non-pitting edema in skin, mucosa, and viscera. Potentially fatal if laryngeal airway is involved. Treatment: recombinant C1-INH, icatibant (bradykinin B2 receptor antagonist), ecallantide (kallikrein inhibitor).
6b. Inhibition of C3/C5 Convertase Assembly - The RCA (Regulators of Complement Activation) Family
All members of the RCA family share complement control protein repeats (CCPRs) - short consensus repeat domains that bind to C3b or C4b.
| Protein | Location | Mechanism | Specificity |
|---|---|---|---|
| DAF (CD55) | Cell membrane (GPI-anchored) | Accelerates decay of C3/C5 convertases; displaces Bb from C3bBb and C2a from C4b2a | Both pathways |
| MCP (CD46) | Cell membrane (transmembrane) | Cofactor for Factor I to cleave C3b → iC3b; also cleaves C4b | Both pathways |
| CR1 (CD35) | Cell membrane (transmembrane) | Cofactor for Factor I; also accelerates convertase decay | Both pathways |
| Factor H | Plasma (glycoprotein) | Competes with Factor B for binding to C3b; displaces Bb from C3bBb; cofactor for Factor I | Alternative pathway only (C3b only) |
| C4BP (C4b-binding protein) | Plasma | Competes with C2 for C4b; cofactor for Factor I-mediated cleavage of C4b | Classical/lectin pathways |
| Factor I | Plasma (serine protease) | Cleaves C3b → iC3b (non-activating) and C4b (non-activating); requires a cofactor (Factor H, MCP, CR1, or C4BP) | Requires cofactor |
PNH (Paroxysmal Nocturnal Hemoglobinuria): Somatic mutation in the PIG-A gene in a hematopoietic stem cell. PIG-A encodes phosphatidylinositol glycan class A - required for GPI-anchor biosynthesis. Loss of GPI anchors causes deficiency of CD55 (DAF) and CD59 on erythrocytes, platelets, and granulocytes. Red cells become hypersensitive to spontaneous complement-mediated lysis → chronic hemolytic anemia. Also causes venous thrombosis (complement activation on platelets). Diagnosed by flow cytometry showing absent CD55/CD59. Treatment: eculizumab (anti-C5 antibody).
6c. Inhibition of MAC Formation
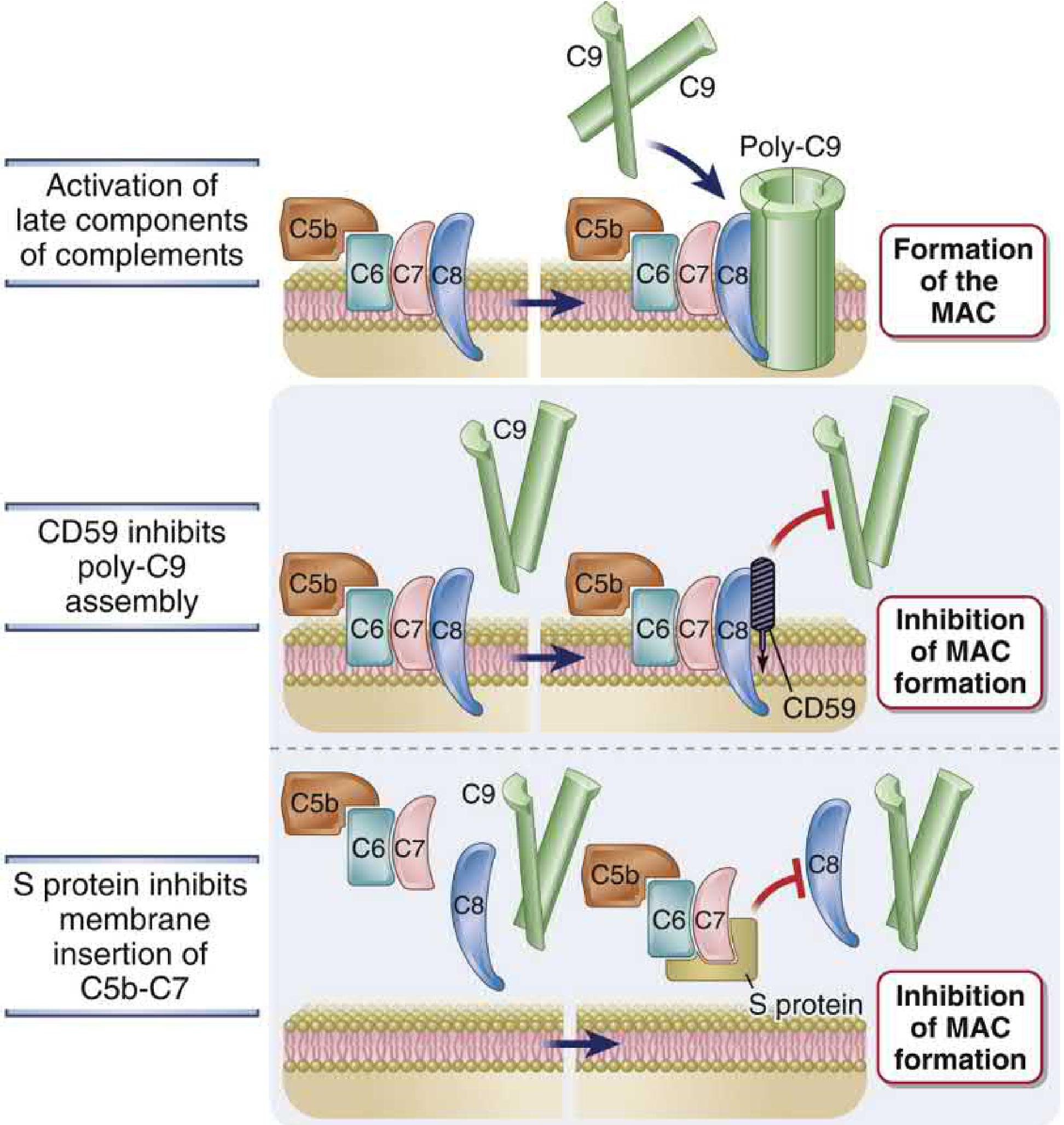
Two mechanisms prevent MAC insertion on host cell membranes:
- CD59 (protectin): GPI-anchored membrane protein on virtually all host cells. Binds to C5b-8 complex and prevents binding and polymerization of C9, thereby blocking MAC formation.
- S protein (vitronectin): Plasma glycoprotein. Binds to hydrophobic C5b-7 complex in the fluid phase, blocking its insertion into host cell membranes. Converts C5b-7 into a soluble, non-lytic complex.
Cellular and Molecular Immunology, pp. 862-869
7. Effector Functions - Detailed
7a. Opsonization and Phagocytosis
The sequence: complement activation → C3b/iC3b deposited on microbe → phagocyte CR1/CR3 binds → phagocytosis.
- C3b (native) binds CR1 on neutrophils and macrophages. CR1 alone does not efficiently trigger phagocytosis unless there is co-stimulation (by IFN-γ or C5a). This two-signal requirement prevents inadvertent phagocytosis of host cells that have small amounts of C3b.
- iC3b (generated by Factor I cleavage of C3b) binds CR3 and CR4, which are more efficient phagocytic receptors.
- When microbes are dual-opsonized (both C3b and IgG), CR1 and FcγR synergize dramatically for phagocytosis and killing.
Clinical importance: Complement-mediated opsonization is the primary defense against encapsulated bacteria (pneumococcus, meningococcus, Haemophilus). IgM antibodies against capsular polysaccharides fix C3b, enabling splenic macrophages to phagocytose and destroy these bacteria. This is why asplenic patients (traumatic splenectomy, sickle cell autosplenectomy) are susceptible to overwhelming post-splenectomy sepsis with these organisms.
7b. Stimulation of Inflammation (Anaphylatoxins)
C3a, C4a, and C5a are "anaphylatoxins" - they trigger reactions characteristic of anaphylaxis:
C5a (most potent):
- Mast cell and basophil degranulation → histamine release → vasodilation, increased permeability
- Neutrophil chemotaxis (the most potent chemoattractant complement fragment)
- Neutrophil activation: firm adhesion to endothelium, respiratory burst (ROS generation), lysosomal enzyme release, upregulation of CR1/CR3
- Direct action on vascular endothelium: P-selectin upregulation, increased permeability
- Activates the lipoxygenase pathway in neutrophils/monocytes → further inflammatory mediator generation
C3a (~20-fold less potent than C5a):
- Mast cell degranulation
- Chemoattractant for eosinophils specifically
C4a (~2500-fold less potent than C5a):
- Binds PAR1/PAR4 receptors; minimal biological effect in humans
Anaphylatoxins are rapidly inactivated by carboxypeptidase N (cleaves the C-terminal arginine from C3a and C5a), generating C3a-desArg and C5a-desArg which are less active.
7c. Complement-Mediated Cytolysis
MAC inserts into the membrane and forms a pore. Net Na⁺ and water influx (driven by high intracellular colloid osmotic pressure) → osmotic lysis. MAC killing is clinically significant mainly against Neisseria (thin cell walls, no peptidoglycan layer preventing access).
Sub-lytic MAC deposition (insufficient to lyse the cell) can still cause:
- Platelet activation and aggregation
- Endothelial cell activation → procoagulant surface → thrombosis
- Smooth muscle cell activation
- Cellular responses including upregulation of inflammatory mediators
7d. Immune Complex Clearance
Complement proteins bind to immune complexes and:
- Sterically block Fc-Fc interactions that promote lattice formation → solubilize immune complexes
- C3b on immune complexes binds to CR1 on erythrocytes → complexes are transported to liver and spleen → stripped and degraded by phagocytes (Kupffer cells)
Failure of this function - as in C1q, C2, or C4 deficiency - leads to accumulation of immune complexes in blood vessel walls → vasculitis, glomerulonephritis → SLE-like disease (>50% of C1q-deficient individuals develop SLE).
7e. Enhancement of Adaptive Humoral Immunity (B Cell Co-receptor)
C3d (the final stable breakdown product of C3b, after Factor I and other proteases act on it) covalently attaches to antigens. B cells can simultaneously:
- Bind antigen through the BCR (Ig receptor)
- Bind C3d on the same antigen through CR2 (CD21)
CR2-CD19-CD81 form the B cell co-receptor complex. Co-engagement of BCR and CR2 by the same antigen lowers the threshold for B cell activation approximately 1000-fold by co-localizing CD19 (which provides powerful signal amplification through PI3K activation) with the BCR.
Opsonized antigens coated with C3d are also captured by follicular dendritic cells (FDCs) (which express CR1 and CR2) in germinal centers of lymphoid organs. FDCs display antigen to germinal center B cells for affinity maturation and selection. Loss of C3, C4, or CR2 in mice dramatically impairs antibody responses and germinal center formation.
7f. Apoptotic Cell Clearance
Complement proteins (especially C1q and C3) bind to apoptotic cells and facilitate their phagocytic removal. This is important to prevent secondary necrosis of apoptotic cells and the release of intracellular self-antigens. Deficiency in this function contributes to SLE pathogenesis.
Cellular and Molecular Immunology, pp. 872-878
8. Complement Deficiency States - Clinical Correlates
| Deficient Component | Key Clinical Manifestations | Mechanism |
|---|---|---|
| C1q, C1r/s, C4, C2 (early classical) | SLE-like disease (>50%); recurrent infections with encapsulated bacteria | Impaired immune complex clearance; impaired opsonization |
| MBL, MASP | Recurrent infections in infancy (before IgG matures); Neisseria | Absent lectin pathway |
| C3 | Most severe: recurrent serious pyogenic infections (pneumococcal sepsis, meningitis); may be fatal | No opsonization, no anaphylatoxins, no MAC |
| Factor D, Factor B, Properdin | Neisseria meningitidis infections; encapsulated bacteria | Loss of alternative pathway amplification |
| C5-C9 (MAC components) | Specific susceptibility to disseminated Neisseria infections | No lytic defense; organisms with thin walls survive |
| C1-INH | Hereditary angioedema (HAE): episodic bradykinin-mediated edema | Uncontrolled C1 activation → C2 kinin + bradykinin |
| Factor H / Factor I | Atypical HUS, C3 glomerulopathy (C3GN), MPGN type II; age-related macular degeneration (Factor H polymorphisms) | Uncontrolled alternative pathway → C3 consumption → deposits |
| DAF/CD59 (GPI-anchor defect - PNH) | Chronic hemolytic anemia, venous thrombosis, cytopenias | Unregulated MAC lysis of RBCs |
| CR3/CR4 (CD18 mutation) | Leukocyte Adhesion Deficiency (LAD): recurrent pyogenic infections, impaired wound healing | Defective neutrophil adhesion to endothelium AND impaired iC3b-dependent phagocytosis |
Complement analysis in clinical practice:
- CH50 (total hemolytic complement): detects deficiency of any classical/terminal pathway component
- AH50: detects alternative pathway deficiency
- C3, C4: consumed in active disease (e.g., lupus nephritis: ↓C3, ↓C4)
- Flow cytometry for CD55/CD59: diagnoses PNH
Henry's Clinical Diagnosis and Management, pp. 1112-1113; Cellular and Molecular Immunology, pp. 875-878
9. Microbial Evasion of Complement
Pathogens have evolved diverse evasion strategies:
Group 1 - Recruiting host regulatory proteins:
- Many pathogens express sialic acids (mimicking host) → recruit Factor H → inhibit alternative pathway. N. gonorrhoeae, Haemophilus, schistosomes enzymatically transfer sialic acids from host to their surfaces. E. coli K1 and some meningococci have their own sialic acid biosynthesis.
- HIV-1 gp41 binds Factor H
- Streptococcus pyogenes M protein binds Factor H and C4BP
- Staphylococcus aureus protein Efb binds C3b; SCIN (Staphylococcal Complement Inhibitor) directly blocks C3 convertases
Group 2 - Physical barriers:
- Thick capsules (gram-positive bacteria, fungi) physically prevent MAC access
- Long polysaccharide chains on gram-negative LPS (smooth LPS) divert MAC away from the outer membrane
Group 3 - Secreting complement-inhibitory proteins:
- Staphylococcal CHIPS (Chemotaxis Inhibitory Protein of Staphylococcus) blocks C5aR1 and FPR1 → inhibits neutrophil chemotaxis
- Herpesvirus proteins (e.g., HCMV UL18, HSV glycoprotein C) block C3b deposition
Cellular and Molecular Immunology, pp. 879-882
10. Therapeutic Targeting of Complement
| Drug | Target | Indications |
|---|---|---|
| Eculizumab (anti-C5 mAb) | Blocks C5 cleavage → no C5a/C5b | PNH, atypical HUS, NMOSD, myasthenia gravis |
| Ravulizumab (long-acting anti-C5) | Same as eculizumab | PNH, aHUS |
| Avacopan (C5aR1 antagonist) | Blocks C5a receptor 1 | ANCA-associated vasculitis |
| Recombinant C1-INH (Berinert, Ruconest) | Replaces deficient C1-INH | Hereditary angioedema |
| Pegcetacoplan (C3 inhibitor) | Blocks C3 cleavage - inhibits all 3 pathways | PNH |
| Danicopan (Factor D inhibitor) | Blocks alternative pathway | PNH breakthrough hemolysis |
The approval of C5 inhibitors (eculizumab) transformed the treatment of PNH and aHUS. However, patients on C5 inhibitors lose their MAC-mediated defense against Neisseria → mandatory meningococcal vaccination before starting therapy.
Summary: Complement System at a Glance
The complement system is one of the most ancient and versatile defense systems in immunity. The three activation pathways (alternative = innate surveillance; lectin = pattern recognition; classical = adaptive effector) all converge on C3b deposition, with three downstream outcomes - opsonization and phagocytosis (via C3b/CR1 and iC3b/CR3), inflammation (via C3a/C5a anaphylatoxins), and lysis (via MAC). Regulatory proteins (C1-INH, DAF, CD59, Factor H, Factor I, MCP) maintain tight control to prevent host-cell damage. Deficiencies reveal the specific roles of each component: C3 deficiency → susceptibility to all pyogenic bacteria; C5-C9 deficiency → specific Neisseria susceptibility; C1-INH deficiency → hereditary angioedema; PIG-A/GPI deficiency → PNH. Modern therapeutics targeting C3, C5, Factor D, and C5aR1 have made complement biology directly clinically actionable.
Sources:
- Cellular and Molecular Immunology (Abbas, Lichtman & Pillai), pp. 838-882
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 97-99
- Janeway's Immunobiology 10e, pp. 1462-1469
- Roitt's Essential Immunology, pp. 850-862
- Henry's Clinical Diagnosis, pp. 1112-1113
only from abbas book
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Here is the complete, detailed account of the complement system drawn exclusively from Abbas, Lichtman & Pillai - Cellular and Molecular Immunology (9th edition).
The Complement System - Abbas (Cellular and Molecular Immunology)
Historical Background
The name "complement" originates from experiments by Jules Bordet shortly after the discovery of antibodies. He demonstrated that fresh serum containing an antibacterial antibody lysed bacteria at 37°C. When the serum was heated to 56°C or more, lytic capacity was lost - but not agglutinating capacity, because antibodies are relatively heat stable. Adding fresh serum from non-immunized animals restored lysis. Bordet concluded that serum contains a heat-labile component present in all individuals that assists, or "complements," the lytic function of antibodies. This component was later named complement.
(Abbas, p. 838)
General Principles of Complement Activation
The complement system consists of serum and cell surface proteins that interact in a highly regulated manner to generate products that eliminate microbes. Complement proteins are plasma proteins that are normally inactive; they are activated only under particular conditions. Several features are essential:
-
Complement attacks microbial surfaces: The system is activated by microbes and by antibodies/lectins bound to microbes. Complement therefore focuses immune attack specifically on microbial surfaces.
-
Proteolytic cascade amplification: Activation involves sequential proteolysis of proteins to generate enzyme complexes. Proteins that acquire proteolytic activity by the action of other proteases are called zymogens. This is also characteristic of the coagulation and kinin systems. Proteolytic cascades allow tremendous and rapid amplification because each active enzyme molecule at one step can generate multiple activated enzyme molecules at the next step.
-
Surface restriction via covalent attachment: Several biologically active cleavage products become covalently attached to microbial cell surfaces, to antibodies bound to microbes and other antigens, and to apoptotic bodies. In the fluid phase, complement proteins are inactive or only transiently active (for seconds), but they become stably activated after attachment to microbes, antibodies, or dying cells. Thus, full activation and biologic functions of the complement system are limited to microbial cell surfaces or to sites of antibody-antigen binding - they do not occur freely in the blood.
-
Self vs. non-self discrimination via regulatory proteins: Complement activation is inhibited by regulatory proteins present on normal host cells but absent from microbes. These regulatory proteins are an adaptation of normal cells that minimize complement-mediated damage. Because microbes lack these proteins, complement activation can proceed on microbial surfaces.
(Abbas, pp. 838-840)
Three Pathways of Complement Activation
There are three pathways: the classical (activated by IgM and IgG bound to antigen), the alternative (activated on microbial cell surfaces in the absence of antibody), and the lectin (activated by plasma lectins binding to surface carbohydrates on microbes). The classical pathway was discovered first, but the alternative pathway is phylogenetically older. The alternative and lectin pathways are effector mechanisms of innate immunity; the classical pathway is a major mechanism of adaptive humoral immunity.
The central event in all three pathways is proteolysis of C3 to generate biologically active products and the covalent attachment of C3b to microbial cell surfaces. All three generate a proteolytic complex called the C3 convertase, which cleaves C3 into C3a and C3b.
(Abbas, pp. 840-842, Fig. 13.6)
The Alternative Pathway
(Abbas, Table 13.4)
| Protein | Structure | Serum Conc. | Function |
|---|---|---|---|
| C3 | 185 kD (α 110 kD, β 75 kD) | 1400-1700 µg/mL | C3b binds microbe surface; opsonin; component of C3 and C5 convertases. C3a is anaphylatoxin |
| Factor B | 93-kD monomer | 200-400 µg/mL | Bb is the serine protease of the alternative C3/C5 convertases |
| Factor D | 25-kD monomer | 1-3 µg/mL | Plasma serine protease; cleaves Factor B when bound to C3b |
| Properdin | Up to four 56-kD subunits | 20-35 µg/mL | Stabilizes C3 convertase (C3bBb) |
The Alternative Pathway - Step by Step
C3 tickover: C3 in plasma is continuously hydrolyzed at a low rate (1%-2% of total plasma C3 per hour) to generate C3b in a process called C3 tickover. The C3 protein contains a reactive thioester bond buried in the thioester domain. When C3 is cleaved, the C3b molecule undergoes a dramatic conformational change and the thioester domain flips out (a shift of approximately 85 Å), exposing the previously hidden reactive thioester bond.
A small amount of C3b may become covalently attached to cell surfaces through the thioester domain, which reacts with amino or hydroxyl groups of cell surface proteins or polysaccharides to form amide or ester bonds. If these bonds are not formed, the exposed reactive thioester bond is quickly hydrolyzed, rendering the protein inactive - so further complement activation cannot proceed in plasma.
On microbial surfaces: Surface-bound C3b binds Factor B. Factor D (a constitutively active serine protease present at very low concentrations) cleaves Factor B in this complex, releasing Ba and generating the C3 convertase: C3bBb. Properdin binds to and stabilizes the C3bBb convertase, significantly prolonging its half-life on microbial surfaces.
Amplification loop: The C3 convertase cleaves more C3 → more C3b deposited covalently on the microbial surface → more C3bBb convertase assembled → more C3 cleaved. This positive feedback loop produces massive amplification.
C5 convertase: When sufficient C3b accumulates, an additional C3b molecule is incorporated into the convertase, forming the C5 convertase: C3bBbC3b, which cleaves C5.
Self/non-self discrimination: On host cells, regulatory proteins (Factor H, DAF, MCP) rapidly inactivate C3b. In addition, cell surfaces rich in sialic acid favor binding of the regulatory protein Factor H over the alternative pathway protein Factor B. Mammalian cells express higher levels of sialic acid than most microbes - another reason complement activation is prevented on normal host cells and permitted on microbes.
(Abbas, pp. 843-845, Fig. 13.7, Table 13.4)
The Classical Pathway
(Abbas, Tables 13.5-13.6)
Structure of C1
C1 is a macromolecular complex consisting of:
- C1q: 6 identical subunits arranged to form a central core with symmetrically projecting radial arms. The globular heads (H) at the end of each arm are the contact regions for the Fc portions of antibodies (IgG or IgM).
- C1r₂s₂: a tetramer of two C1r and two C1s molecules that wraps around the radial arms of C1q, juxtaposing the catalytic domains of C1r and C1s.
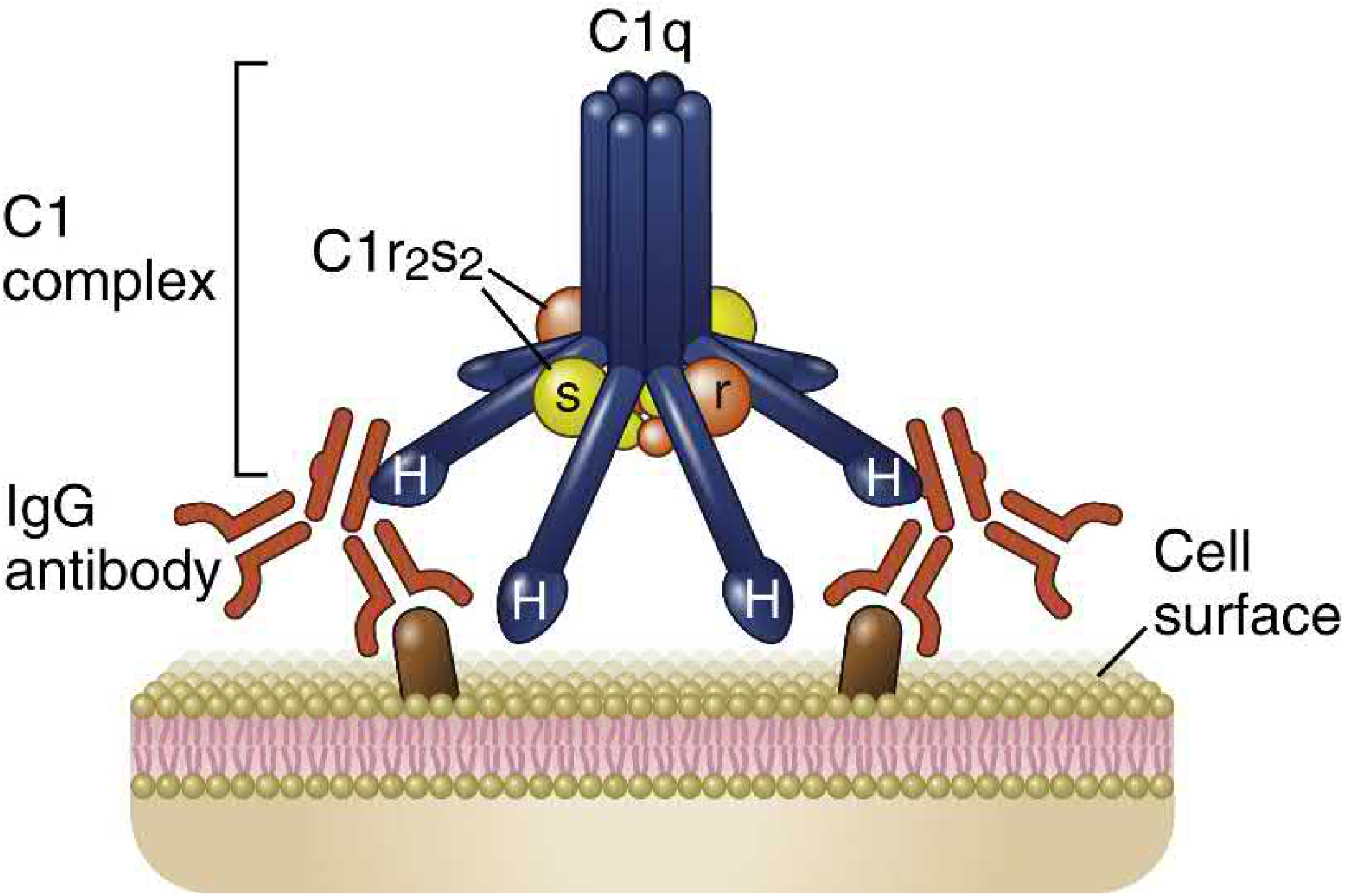
(Abbas, p. 854, Fig. 13.10)
Initiation
C1q binds to the Fc regions (CH2 domain) of IgG or IgM that have bound to antigen. For activation, at least two C1q globular heads must simultaneously engage Ig Fc regions. This explains the difference between antibody isotypes:
- IgM (pentamer): highly efficient - one IgM molecule provides up to 5 potential Fc sites in close proximity. Even a single IgM molecule bound to antigen can activate complement.
- IgG (monomer): requires two adjacent IgG molecules bound to antigen. Soluble IgG does not activate the classical pathway.
Downstream Steps
C1q binding induces a conformational change → C1r autoactivates → C1r cleaves and activates C1s (a serine protease). Active C1s sequentially cleaves:
- C4 → C4a (small, released, weak anaphylatoxin) + C4b (large, covalently attaches to surface via thioester bond)
- C2 → C2b (released) + C2a (remains associated with C4b)
Classical pathway C3 convertase: C4b2a
This then cleaves C3 to generate C3a and C3b. C3b joins the convertase to form the classical C5 convertase: C4b2aC3b.
Note: Pentraxins (such as C-reactive protein, an acute-phase protein) can also bind C1q and initiate the classical pathway, linking acute-phase responses to complement.
(Abbas, pp. 846-852)
The Lectin Pathway
Mannose-binding lectin (MBL) is a plasma protein that recognizes terminal mannose residues on microbial glycoproteins and glycolipids. MBL is a member of the collectin family with a hexameric structure similar to C1q.
After MBL binds to microbes, two zymogens called MASP1 (mannose-binding lectin-associated serine protease 1) and MASP2 - with functions similar to those of C1r and C1s - associate with MBL and initiate downstream proteolytic steps identical to the classical pathway.
The same C3 convertase (C4b2a) and same C5 convertase (C4b2aC3b) are generated. The subsequent late steps are identical to the classical pathway.
(Abbas, pp. 852-853)
Late Steps: C5 Cleavage and the Membrane Attack Complex (MAC)
C5 convertases generated by any of the three pathways initiate activation of the late components, culminating in the MAC.
(Abbas, Table 13.7)
| Protein | Structure | Serum Conc. | Function |
|---|---|---|---|
| C5 | 190-kD dimer (115-kD + 75-kD chains) | 80 µg/mL | C5b initiates MAC assembly; C5a stimulates inflammation |
| C6 | 110-kD monomer | 45 µg/mL | Binds C5b; accepts C7 |
| C7 | 100-kD monomer | 90 µg/mL | Binds C5b,6; inserts into lipid membranes |
| C8 | 155-kD trimer (64 + 64 + 22-kD chains) | 60 µg/mL | Binds C5b,6,7; initiates binding and polymerization of C9 |
| C9 | 79-kD monomer | 60 µg/mL | Binds C5b,6,7,8; polymerizes to form membrane pores |
MAC Assembly - Step by Step
C5 convertase cleaves C5:
- C5a (small, released): potent pro-inflammatory fragment
- C5b (large): released but rapidly binds plasma C6, undergoing a conformational change
The C5b-C6 complex binds to the cell membrane through ionic and hydrophobic interactions. C7 then binds to the α chain of C5b, forming C5b-7, which undergoes an amphiphilic transition and penetrates the membrane (can release phospholipid micelles but does not yet form complete pores).
C8 (a trimer: one chain binds C5b; a second chain forms a covalent heterodimer with it; the third chain inserts into the lipid bilayer). The stably inserted C5b-8 complex forms unstable pores ranging from 0.4 to 3 nm in diameter - sufficient for lysis in very large numbers.
C9 (the final complement component) polymerizes at the C5b-8 site to form pores made up of C5b-9 complexes containing C5b, C6, C7, C8, and many molecules of C9. These pores are:
- ~20 nm external diameter
- 1-11 nm internal diameter (varies with number of C9 molecules)
- ~15 nm height
- Fully permeable to water and ions
The channel size varies based on the number of C9 molecules in the C5b-C9 complex. Tubular complexes of C9 alone may also form. The entry of water results in osmotic swelling and rupture of the cells on whose surface the MAC is deposited.
Abbas explicitly notes: C9 is structurally homologous to perforin - the cytolytic granule protein of cytotoxic T lymphocytes and NK cells - illustrating evolutionary conservation of this membrane-disruption mechanism.
(Abbas, pp. 853-858, Fig. 13.12, Table 13.7)
Receptors for Complement Proteins
(Abbas, Table 13.8, pp. 858-862)
CR1 (CD35) - Type 1 Complement Receptor
Ligands: C3b, C4b (high-affinity)
Distribution: Erythrocytes, neutrophils, monocytes, macrophages, eosinophils, T and B lymphocytes, follicular dendritic cells (FDCs)
Functions:
- Promotes phagocytosis of C3b- and C4b-coated particles. The binding of C3b- or C4b-coated particles to CR1 transduces signals that activate microbicidal mechanisms of phagocytes, especially when the Fcγ receptor is simultaneously engaged by antibody-coated particles (dual opsonization).
- Immune complex clearance: CR1 on erythrocytes binds circulating immune complexes with attached C3b/C4b and transports them to the liver and spleen. Phagocytes there remove the immune complexes from the erythrocyte surface, and the erythrocytes continue to circulate.
- Complement regulation: CR1 is also a regulator of complement activation (cofactor for Factor I).
CR2 (CD21) - Type 2 Complement Receptor
Ligands: C3d, C3dg, iC3b (cleavage products of C3b generated by Factor I)
Distribution: B lymphocytes, FDCs, some epithelial cells
Functions:
- B cell coreceptor: CR2 is expressed on mature B cells as part of a trimolecular complex with CD19 and CD81 (TAPA1). This complex delivers signals that enhance B cell responses to antigen (detailed below).
- Antigen trapping on FDCs: CR2 on FDCs traps iC3b-, C3d-, and C3dg-coated antigen-antibody complexes in germinal centers.
- CR2 is also the receptor for Epstein-Barr virus (EBV), which uses it to infect B cells.
CR3 (MAC-1, CD11b:CD18) - Type 3 Complement Receptor
Ligand: iC3b (the inactive fragment generated by Factor I proteolysis of C3b)
Distribution: Neutrophils, mononuclear phagocytes, mast cells, NK cells
Functions:
- Promotes phagocytosis of microbes opsonized with iC3b.
- May directly recognize some microbial molecules for phagocytosis.
- Binds ICAM-1 on endothelial cells and promotes stable attachment of leukocytes to endothelium - leading to recruitment of leukocytes to sites of infection and tissue injury.
CR3 is a member of the integrin family: its α chain (CD11b) is noncovalently linked to a β chain (CD18) that is shared with two closely related integrin molecules - LFA-1 and p150,95 (CR4). Deficiency of CD18 (the shared β chain) causes Leukocyte Adhesion Deficiency (LAD), characterized by recurrent pyogenic infections.
CR4 (CD11c:CD18)
Ligand: iC3b
Distribution: Macrophages, monocytes, neutrophils, dendritic cells
Function: Phagocytosis of iC3b-opsonized particles (shares the CD18 β chain with CR3).
(Abbas, pp. 858-862, Table 13.8)
Regulation of Complement Activation
(Abbas, Table 13.9, pp. 862-870)
Activation of the complement cascade and the stability of active complement proteins are tightly regulated to prevent complement activation on normal host cells and to limit the duration of active complexes. Regulation is mediated by circulating and cell membrane proteins, many of which belong to the family called Regulators of Complement Activity (RCA), encoded by homologous genes located on chromosome 1q3.2.
RCA proteins include membrane proteins DAF (CD55), MCP (CD46), CR1, CR2, and plasma proteins Factor H and C4-binding protein (C4BP).
Complement activation must be regulated for two reasons:
- Low-level complement activation occurs spontaneously, and if allowed to proceed, it can damage normal cells and tissues.
- Even when appropriately activated on microbial cells, degradation products can diffuse to adjacent host cells and injure them.
Regulatory Proteins Table (from Abbas Table 13.9)
| Regulator | Location | Interacts With | Function |
|---|---|---|---|
| C1 inhibitor (C1-INH) | Plasma (104 kD); 200 µg/mL | C1r, C1s | Serine protease inhibitor (serpin); binds and dissociates C1r and C1s from C1q; stops classical and lectin pathway activation |
| Factor I | Plasma (88-kD dimer); 35 µg/mL | C4b, C3b | Serine protease; cleaves C3b and C4b using Factor H, MCP, C4BP, or CR1 as cofactors; converts C3b → iC3b |
| Factor H | Plasma (150 kD, multiple CCPRs); 480 µg/mL | C3b | Binds C3b and displaces Bb (alternative pathway); cofactor for Factor I |
| C4-binding protein (C4BP) | Plasma (570 kD, multiple CCPRs); 300 µg/mL | C4b | Cofactor for Factor I-mediated cleavage of C3b; competes with C2 for C4b binding |
| MCP (CD46) | Membrane (45-70 kD, transmembrane); leukocytes, epithelial/endothelial cells | C3b, C4b | Binds C4b and displaces C2; cofactor for Factor I-mediated cleavage of C3b and C4b |
| DAF (CD55) | Membrane (70 kD, GPI-linked, four CCPRs); blood cells, endothelial, epithelial | C4b2a, C3bBb | Displaces C2a from C4b and Bb from C3b - dissociation of both C3 convertases |
| CD59 | Membrane (18 kD, GPI-linked); blood cells, endothelial, epithelial | C7, C8 | Inhibits MAC formation - blocks C9 polymerization |
Detailed Regulatory Mechanisms
1. Inhibition of C1 Activation - C1-INH
C1-INH is a serpin (serine protease inhibitor) that mimics the normal substrates of C1r and C1s. It becomes a "suicide substrate" - cleaved by and covalently attached to C1r and C1s, causing the C1r₂-C1s₂ tetramer to dissociate from C1q, stopping classical pathway activation. By also inactivating MASP2, C1-INH dampens the lectin pathway. C1-INH also inhibits kallikrein and Factor XII, limiting bradykinin production.
Hereditary angioedema (HAE): Autosomal dominant deficiency of C1-INH. Uncontrolled C1 activation → increased breakdown of C4 and C2 → generation of C2 kinin and bradykinin → episodic accumulation of edema fluid in skin and mucosa → abdominal pain, vomiting, diarrhea, and potentially life-threatening airway obstruction. Treatment: recombinant C1-INH.
2. Inhibition of C3/C5 Convertase Assembly - The RCA Family
If C3b deposits on the surface of normal mammalian cells, it is bound by membrane proteins (MCP, CR1, DAF) and the plasma protein Factor H. C4b deposited on cell surfaces is similarly bound by DAF, CR1, MCP, and C4BP. By binding C3b or C4b, these proteins competitively inhibit binding of other convertase components (Bb of the alternative pathway or C2a of the classical pathway), blocking further cascade progression.
- Factor H inhibits binding of only Bb to C3b → regulates the alternative pathway only (not the classical pathway)
- MCP, CR1, and DAF are produced by mammalian cells but not microbes → selectively inhibit complement on host cells while permitting activation on microbes
- Sialic acid on mammalian cells (higher than on microbes) further favors binding of Factor H over Factor B
DAF and PNH: DAF is a GPI-linked membrane protein on endothelial cells and erythrocytes. A hematopoietic stem cell deficiency of the enzyme required to synthesize GPI (encoded by the PIG-A gene) leads to Paroxysmal Nocturnal Hemoglobinuria (PNH): loss of DAF and CD59 from blood cells → excessive complement activation → chronic hemolysis, thrombosis.
3. Inhibition of MAC Formation
The MAC inhibitors prevent self-cell destruction:
- CD59 (protectin): GPI-linked membrane protein. Binds to C5b-8 complex and prevents binding and polymerization of C9, blocking MAC formation.
- S protein (vitronectin): Plasma protein. Binds to hydrophobic C5b-7 in the fluid phase, blocking its membrane insertion.
(Abbas, pp. 862-870, Figs. 13.13-13.16, Table 13.9)
Functions of the Complement System
(Abbas, pp. 870-878, Fig. 13.17)
The principal functions are: (1) promote phagocytosis, (2) stimulate inflammation, and (3) induce lysis. In addition, complement promotes B lymphocyte activation and humoral immune responses.
1. Opsonization and Phagocytosis
Microbes coated with C3b, iC3b, or C4b are phagocytosed by binding to specific receptors on macrophages and neutrophils. C3b and C4b bind CR1; iC3b binds CR3 (Mac-1) and CR4.
CR1 alone is inefficient at inducing phagocytosis of C3b-coated microbes. Its ability is enhanced when:
- Microbes are simultaneously coated with IgG (Fcγ receptor and CR1 co-engagement)
- Macrophages are activated by the cytokine IFN-γ
Clinical importance: The clearest example is host defense against encapsulated bacteria (pneumococci, meningococci). IgM antibodies against capsular polysaccharides fix C3b → phagocytic clearance by the spleen. This is why asplenic individuals (autosplenectomy in sickle cell anemia, surgical removal, traumatic rupture) are susceptible to overwhelming post-splenectomy sepsis with these organisms.
2. Stimulation of Inflammatory Responses (Anaphylatoxins)
C5a, C4a, and C3a induce acute inflammation by activating mast cells, neutrophils, and endothelial cells:
- All three peptides bind mast cells and induce degranulation → release of vasoactive mediators (histamine) → vasodilation and increased vascular permeability
- They are called anaphylatoxins because mast cell reactions they trigger are characteristic of anaphylaxis
C5a (the most potent):
- Stimulates neutrophil motility
- Stimulates firm adhesion of neutrophils to endothelium
- At high doses, stimulates the respiratory burst and production of reactive oxygen species
- Acts directly on vascular endothelial cells → increased vascular permeability and expression of P-selectin → promotes neutrophil binding
- These combined actions on mast cells, neutrophils, and endothelial cells contribute to inflammation at sites of complement activation
Relative potency: C5a is most potent; C3a is approximately 20-fold less potent than C5a; C4a is approximately 2500-fold less potent than C5a.
3. Complement-Mediated Cytolysis
Complement-mediated lysis of foreign organisms is mediated by the MAC. Most pathogens have evolved thick cell walls or capsules that impede MAC access. Complement-mediated lysis is critical for defense against only a few pathogens that are unable to resist MAC insertion - specifically bacteria of the genus Neisseria, which have very thin cell walls. This explains why deficiency of terminal complement components (C5-C9) gives specific susceptibility to disseminated Neisseria infections.
4. Functions in Humoral Immunity - B Cell Co-receptor
The C3d protein (generated by Factor I and other proteases acting on C3b) remains covalently attached to the antigen. B lymphocytes can simultaneously:
- Bind antigen via their membrane Ig (BCR)
- Bind attached C3d via CR2 (CD21)
This simultaneous engagement brings CD19 (part of the CR2-CD19-CD81 coreceptor complex) into proximity with BCR-associated kinases. The cytoplasmic tail of CD19 becomes rapidly tyrosine phosphorylated, leading to activation of PI3-kinase → generates PIP3 → activates BTK and PLCγ2. The net result is that the response of the antigen-stimulated B cell is greatly enhanced.
Opsonized antigens coated with C3d are also bound by follicular dendritic cells (FDCs) in germinal centers (via FDC-expressed CR2). FDCs display antigens to germinal center B cells, which is important for selection of high-affinity B cells (affinity maturation). The importance of complement in humoral responses is illustrated by impairment in antibody production and germinal center formation seen in knockout mice lacking C3, C4, or CR2.
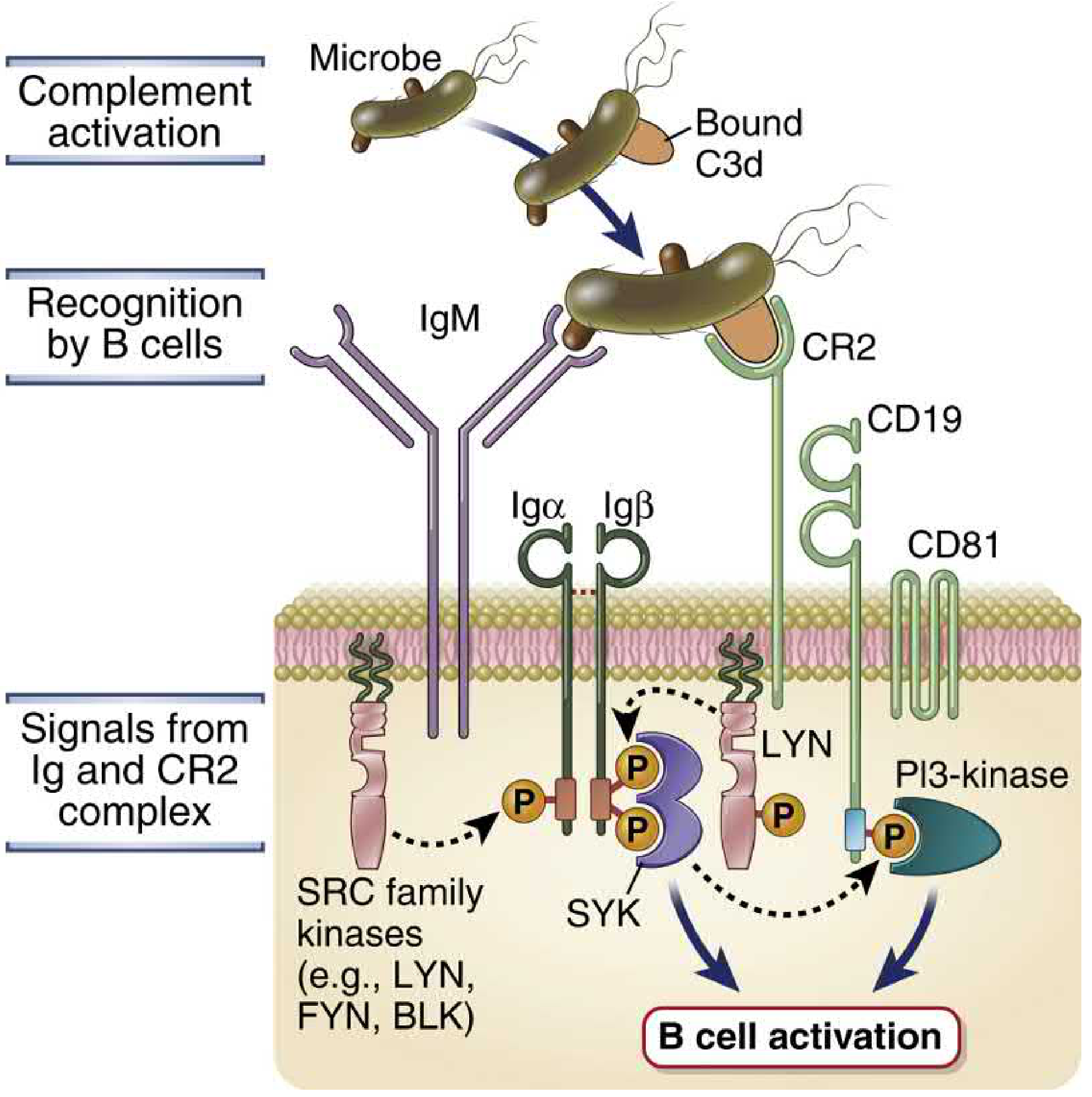
(Abbas, pp. 511-512, Fig. 7.20; pp. 872-875)
5. Immune Complex Clearance and Solubilization
Complement proteins bound to immune complexes:
- Sterically block Fc-Fc interactions that promote lattice formation → solubilize immune complexes
- C3b on immune complexes binds to CR1 on erythrocytes → complexes transported to liver and spleen → phagocytes remove the complexes, erythrocytes continue to circulate
6. Clearance of Apoptotic Cells
Complement proteins (especially C1q and C3) bind to apoptotic cells and facilitate their phagocytic removal, preventing secondary necrosis and the release of intracellular self-antigens. This function explains the link between early complement deficiencies and SLE.
(Abbas, pp. 872-878)
Complement Deficiencies
(Abbas, pp. 874-878)
| Deficiency | Clinical Manifestation | Mechanism |
|---|---|---|
| C1q, C1r, C4, C2 (early classical) | >50% develop SLE-like disease; recurrent infections | Impaired immune complex clearance; impaired C3b opsonization; failed apoptotic cell clearance → self-antigen presentation |
| C2 | Most common human complement deficiency; SLE risk; some asymptomatic | C2 required for C3 convertase (C4b2a) |
| C3 | Frequent serious pyogenic bacterial infections, often fatal; most severe complement deficiency | C3 central to all three pathways: no opsonization, no anaphylatoxins, no MAC initiation |
| Factor B, Factor D (alternative pathway) | Susceptibility to meningococcal infections; rare | Loss of alternative pathway |
| Properdin | Meningococcal infections; X-linked recessive | Loss of C3 convertase stabilization |
| MBL, MASP2 | Recurrent infections especially in infancy | Absent lectin pathway |
| C5, C6, C7, C8, C9 (terminal/MAC) | Specific susceptibility to disseminated Neisseria (N. meningitidis, N. gonorrhoeae); no other consistent problem | MAC required for killing thin-walled organisms |
| C1-INH | Hereditary angioedema (HAE): episodic edema in skin, mucosa, viscera; potentially fatal laryngeal obstruction | Uncontrolled C1 → C2 kinin + bradykinin |
| DAF (CD55) - via PIG-A mutation | PNH: chronic hemolytic anemia, venous thrombosis | GPI-anchor loss → no DAF/CD59 on RBCs |
| Factor I | Depleted plasma C3 (unregulated tickover C3 convertase); pyogenic bacterial infections | No C3b inactivation → continuous C3 consumption |
| Factor H | Excess alternative pathway activation; C3 consumption; glomerulonephritis; atypical HUS | Mutant Factor H binds poorly to C3b on endothelial surfaces → uncontrolled complement on endothelium → microthrombi and vascular damage |
| CR3/CR4 (CD18 deficiency) | Leukocyte Adhesion Deficiency (LAD): recurrent pyogenic infections; impaired wound healing | Defective neutrophil adhesion to endothelium AND impaired iC3b-mediated phagocytosis |
Atypical HUS and Factor H: Abbas specifically describes this: children present with microangiopathic hemolytic anemia, thrombocytopenia, and acute renal failure triggered by endothelial cell injury caused by hyperactivation of the alternative pathway. Mutant Factor H or MCP binds less well to C3b and C4b on endothelial surfaces → excessive complement activation → formation of microthrombi and vascular damage.
(Abbas, pp. 874-878)
Pathologic Effects of Complement
(Abbas, pp. 878-879)
Even when properly regulated, complement can cause significant tissue damage:
-
Complement activation by antibodies against vascularized organ transplants and immune complexes in autoimmune diseases may bind to vascular endothelium → inflammation → MAC damage to endothelial surface → favors coagulation and intravascular thrombosis → ischemic injury.
-
Some late complement proteins may activate prothrombinases in the circulation → thrombosis independent of MAC-mediated endothelial damage.
-
Immune complex-mediated diseases: Systemic vasculitis and immune complex glomerulonephritis result from deposition of antigen-antibody complexes in vessel walls and kidney glomeruli. Complement activated by these complexes initiates acute inflammatory responses that destroy vessel walls or glomeruli → thrombosis, ischemic damage, scarring.
-
The function of regulatory proteins may be overwhelmed when large amounts of antibodies deposit on host cells, generating enough active complement proteins that regulatory molecules are unable to keep up.
Therapeutic targeting: Abbas notes two approved agents:
- Antibodies against C5 that block proteolytic cleavage of C5 → used for PNH, complement-mediated HUS, and neuromyelitis optica
- Recombinant human C1-INH → used for hereditary angioedema
(Abbas, pp. 878-879)
Evasion of Complement by Microbes
(Abbas, pp. 879-882)
Evasion mechanisms fall into three groups:
Group 1 - Recruiting Host Complement Regulatory Proteins
Many pathogens express sialic acids, which inhibit the alternative pathway by recruiting Factor H (which displaces C3b from Bb):
- Schistosomes, N. gonorrhoeae, and certain Haemophilus species scavenge sialic acids from the host and enzymatically transfer them to their cell surfaces
- E. coli K1 and some meningococci have evolved their own biosynthetic routes for sialic acid generation
- HIV gp41 binds Factor H → virion protection
- HIV also incorporates GPI-anchored host regulatory proteins DAF and CD59 into its envelope when budding from infected cells
- S. pyogenes, B. burgdorferi, N. gonorrhoeae, N. meningitidis, Candida albicans, and Echinococcus granulosus synthesize proteins that recruit Factor H to their cell walls
Group 2 - Producing Proteins That Inhibit Complement Steps
- E. coli makes a C1q-binding protein that inhibits association of C1q, C1r, and C1s
- S. aureus makes Staphylococcal Complement Inhibitor (SCIN) that binds to and stably inhibits both classical and alternative pathway C3 convertases → inhibits all three complement pathways
- Herpes simplex virus glycoprotein C destabilizes the alternative pathway convertase by preventing its C3b from binding to properdin
- Trypanosoma cruzi GP160 binds C3b, prevents C3 convertase formation, and accelerates its decay
- Vaccinia virus complement inhibitory protein structurally resembles human C4BP but binds both C4b and C3b → accelerates decay of both C3 and C5 convertases
Group 3 - Inhibiting Complement-Mediated Inflammation
- S. aureus synthesizes CHIPS (Chemokine Inhibitory Protein of Staphylococci) - an antagonist of the C5a anaphylatoxin, blocking neutrophil chemotaxis
(Abbas, pp. 879-882)
Summary (as stated in Abbas)
The antibody effector functions mediated via complement include: blocking infectivity (neutralization), opsonization for phagocytosis (via C3b and Fcγ receptors), activation of NK cells and complement-mediated lysis. Complement is activated by the classical pathway (IgM, IgG), the alternative pathway (microbial surfaces), and the lectin pathway (mannose on microbes), all converging on C3b deposition. C3b is the central opsonin; anaphylatoxins (C3a, C5a) drive inflammation; the MAC (C5b-9) lyses thin-walled organisms. Regulatory proteins protect host cells. Deficiencies expose the specific role of each component.
Source: Abbas, Lichtman & Pillai - Cellular and Molecular Immunology (9th edition, ISBN 9780323757485), Chapter 13 (pp. 838-882) and Chapter 7 (pp. 511-512)
then
Asking for Preferences
This is a shared conversation. Sign in to Orris to start your own chat.