Hb electrophoresis
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Finding Sources
I now have comprehensive information to write a detailed, well-illustrated answer.
Hemoglobin Electrophoresis
Hemoglobin electrophoresis is the separation of hemoglobin (Hb) variants based on their molecular charge and size as they migrate through a gel or medium under an electric field. It is the gold standard for identifying normal and abnormal hemoglobin types and is a core component of newborn screening programs.
Principle
Different hemoglobin molecules carry different net charges depending on their amino acid composition. In an electric field, they migrate at different speeds and distances, producing distinct bands that can be identified by their position.
Methods
1. Alkaline Electrophoresis (pH 8.6) - Cellulose Acetate
This is the routine/primary method.
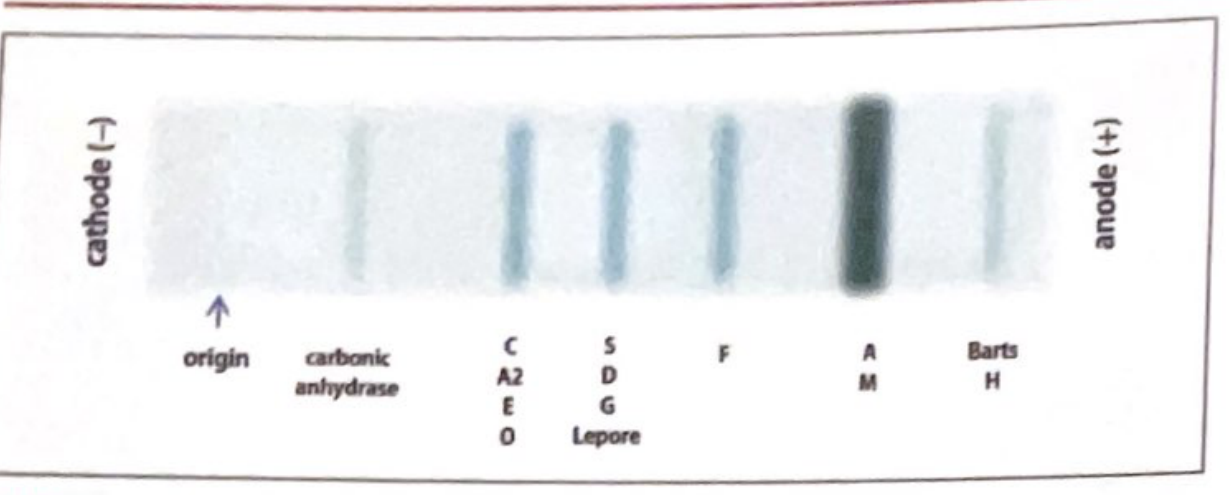
Migration order (cathode → anode):
| Band Position | Hemoglobins Present |
|---|---|
| Origin | - |
| Carbonic anhydrase (reference) | - |
| C band | HbC, HbA2, HbE, HbO |
| S band | HbS, HbD, HbG, Hb-Lepore |
| F band | HbF |
| A band (fastest/major) | HbA, HbM |
| Fast (beyond A) | Hb Barts, HbH |
Normal adult pattern: >97% HbA, <3% HbA2, nothing else.
Key limitation: Cannot distinguish HbS from HbD, HbG, or Hb-Lepore on this method alone.
2. Acid Electrophoresis (pH 6.2) - Citrate Agar
Used as a confirmatory/complementary method when alkaline electrophoresis is uncertain.
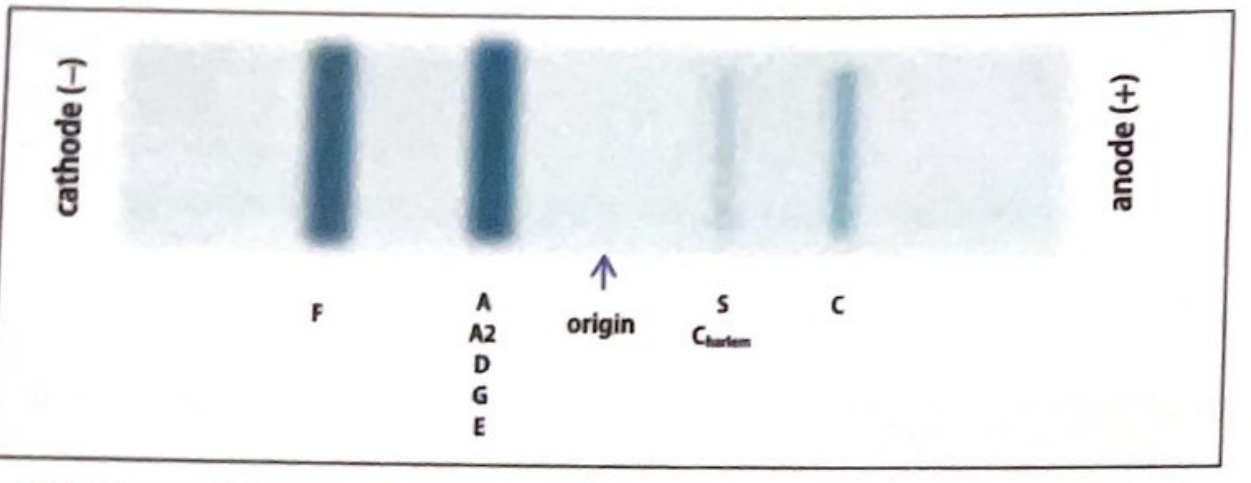
On acid gel, the migration pattern reverses for several variants:
| Band Position | Hemoglobins |
|---|---|
| Near cathode | HbF, HbA/A2/D/G/E |
| Mid-post origin | HbS, HbC-Harlem |
| Near anode | HbC |
Key use: Differentiates HbS from HbD and HbG (which co-migrate on alkaline but separate on acid).
Limitation: Still does not separate HbD from HbG/Hb-Lepore, or HbE from HbO-Arab.
3. HPLC (High Performance Liquid Chromatography)
The preferred modern method - molecules elute at different, characteristic retention times.
Advantages over electrophoresis:
- Accurately quantifies HbA2 and HbF (electrophoresis cannot do this reliably)
- Identifies and separates most variants
Limitations:
- HbE and HbA2 have similar retention times (hard to separate)
- HbC and HbO-Arab have similar retention times
- Bilirubin co-elutes with Hb Barts
4. Capillary Electrophoresis
- Accurately quantifies low HbA2 levels
- Separately quantifies HbE levels
- Note: HbC still runs with HbA2 on this method
5. Isoelectric Focusing (IEF)
Used especially in newborn screening programs - can detect HbS in neonates; often used alongside HPLC.
6. Molecular Methods (PCR/Gene Sequencing)
~1-2% of Hb variants detected by HPLC or electrophoresis cannot be definitively identified and require gene sequencing.
Interpreting Results - Common Patterns
Normal Adults
| Hb Fraction | % |
|---|---|
| HbA | >97% |
| HbA2 | <3% |
| HbF | <1% (trace) |
Sickle Cell Disease States (HbS-related)
| Condition | Electrophoresis Pattern |
|---|---|
| Sickle cell trait (HbAS) | HbA ~58%, HbS ~39%, HbA2 ~2%, HbF ~1% |
| Sickle cell anemia (HbSS) | HbS predominant, elevated HbF, absent/minimal HbA |
| HbSC disease | HbS + HbC, no HbA |
| HbS-β⁰ thalassemia | HbS predominant, elevated HbA2 (>3.5%), no HbA |
| HbS-β⁺ thalassemia | HbS major, HbA small (5-25%), elevated HbA2 + HbF |
Thalassemia
| Condition | Key Finding |
|---|---|
| β-thalassemia trait | HbA2 >3.5% (hallmark finding) + thalassemic indices (low MCV, high RBC count) |
| β-thalassemia major | Predominantly HbF, markedly reduced or absent HbA |
| α-thalassemia | Thalassemic indices but normal HbA2 (cannot diagnose by electrophoresis alone - need HPLC or DNA studies) |
Pitfall: In α-thalassemia co-inherited with β-thalassemia trait, the MCV may normalize and HbA2 levels may be falsely lowered, masking the diagnosis.
Neonatal Hemoglobin Electrophoresis Patterns
Reported in order of decreasing abundance (e.g., FA = more HbF than HbA):
| Pattern | Interpretation |
|---|---|
| FA | Normal newborn (HbF + HbA) |
| FAS | Sickle cell trait (benign) |
| FS | HbSS or HbS-β⁰ thalassemia - clinically significant |
| FSC | HbSC disease - clinically significant |
| FSA | HbS-β⁺ thalassemia - clinically significant |
| FAC | HbC trait (benign) |
| FC | Homozygous HbCC (mild disorder) |
| F only | β-thalassemia major or HPFH (hereditary persistence of HbF) |
| AF | Likely prior blood transfusion - repeat at 4 months |
| FAV / FV | Anomalous band present - repeat/further workup |
Clinical Indications
- Screening for sickle cell disease (mandatory newborn screening in many countries)
- Diagnosis of thalassemia syndromes
- Confirmation of a positive Sickledex/metabisulfite sickle prep (these screening tests must always be confirmed with electrophoresis)
- Carrier detection for genetic counseling
- Evaluation of hemolytic anemia of unknown cause
- Screening in at-risk ethnic populations (e.g., African, Mediterranean, Indian, Southeast Asian ancestry)
- Pre-operative workup (e.g., before anesthesia in at-risk patients)
- Maternal-fetal medicine: screening pregnant women at risk for hemoglobinopathy
Summary Comparison Table
| Method | pH | Best For | Key Limitation |
|---|---|---|---|
| Alkaline electrophoresis | 8.6 | Initial screening, band identification | Cannot separate HbS/D/G/Lepore |
| Acid electrophoresis | 6.2 | Confirming S vs D/G | Cannot separate D/G/Lepore; or E/O-Arab |
| HPLC | - | Quantification of HbA2, HbF | HbE/A2 and HbC/O-Arab overlap |
| Capillary electrophoresis | - | Quantifying low HbA2 | HbC runs with HbA2 |
| IEF | - | Newborn screening | - |
| PCR/Sequencing | - | Definitive variant ID | Resource-intensive |
Sources: Quick Compendium of Clinical Pathology, 5th ed. | The Harriet Lane Handbook, 23rd ed. (Johns Hopkins Hospital) | Creasy & Resnik's Maternal-Fetal Medicine | Henry's Clinical Diagnosis and Management by Laboratory Methods
Paroxysmal nocturnal hemoglobinuria
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Reading File
I now have comprehensive, well-sourced information with excellent diagrams. Here is the full answer:
Paroxysmal Nocturnal Hemoglobinuria (PNH)
PNH is an acquired clonal stem cell disorder - the only hemolytic anemia caused by an acquired (somatic) genetic defect. Despite its name, paroxysmal nocturnal hemoglobinuria is present in only ~25% of cases; most patients have chronic, continuous hemolysis without dramatic hemoglobinuria.
Incidence: ~2-5 per million per year (rare but clinically important)
Pathophysiology
The Genetic Defect
A somatic mutation in the PIGA gene (phosphatidylinositol glycan complementation group A) occurs in a hematopoietic stem cell. PIGA is X-linked, so a single mutation in the active PIGA allele is sufficient to produce the deficiency. Because it arises in a stem cell, all clonal progeny - red cells, white cells, and platelets - are affected.
The Molecular Defect
PIGA encodes an enzyme essential for synthesizing GPI (glycosylphosphatidylinositol) anchors - the lipid structures that attach certain proteins to the outer cell membrane. Without GPI anchors, multiple complement-regulatory proteins are absent from the cell surface.
Missing GPI-Linked Complement Regulatory Proteins
| Protein | Alias | Normal Function |
|---|---|---|
| CD55 | DAF (Decay Accelerating Factor) | Breaks down C3 and C5 convertases; inhibits complement amplification |
| CD59 | MIRL (Membrane Inhibitor of Reactive Lysis) | Blocks C5b-9 MAC assembly - the most important missing protein |
| C8-binding protein | Homologous restriction factor | Inhibits MAC formation |
Other deficient proteins include CD58, CD14, CD24, CD16a, acetylcholinesterase, and leukocyte alkaline phosphatase.
How Hemolysis Occurs
Without CD55 and CD59, the complement cascade proceeds unchecked on the RBC surface, culminating in the C5b-9 Membrane Attack Complex (MAC), which punches holes in the RBC membrane causing intravascular hemolysis.
Why nocturnal? During sleep, there is a slight drop in blood pH, which increases complement activity. However, this explains only a minority of cases - hemosiderinuria (from chronic low-level hemolysis) is almost always present even without visible hemoglobinuria.
Why paroxysmal? Bouts of hemolysis can be triggered by:
- Infection
- Surgery
- Blood transfusion (whole blood)
- Contrast dye injection
- Severe exercise
- Certain medications
Three Types of PNH Red Cells (by complement sensitivity)
| Type | Sensitivity to Lysis | Mechanism |
|---|---|---|
| Type I | Normal | Full GPI-linked proteins present |
| Type II | 3-5x normal | Partial GPI deficiency |
| Type III | 15-25x normal | Complete GPI deficiency |
Complement Pathway and Drug Targets
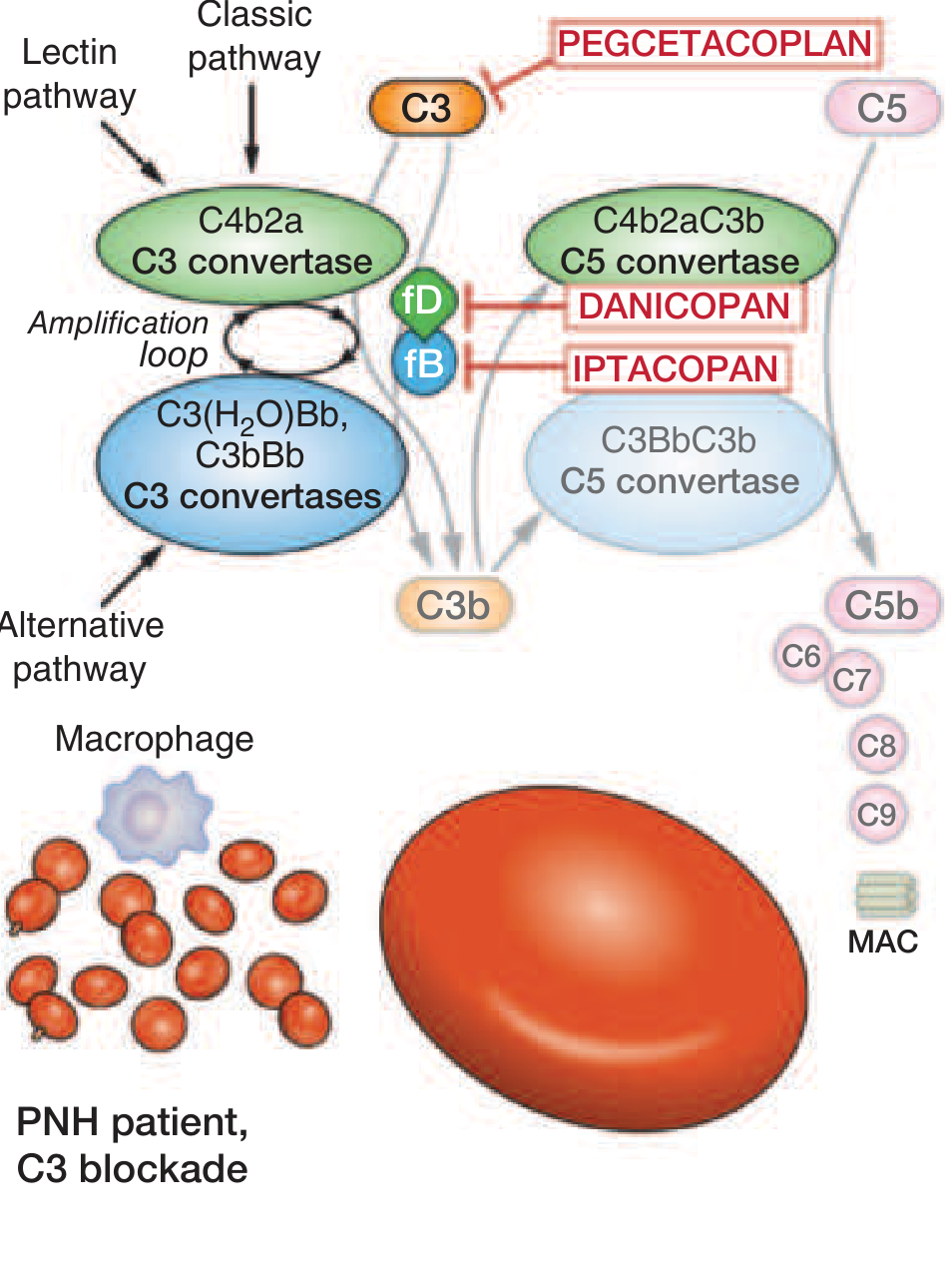
All three complement pathways (classic, lectin, alternative) converge to activate C3 convertase, which generates C3b, then C5 convertase, which cleaves C5 to C5b, assembling the MAC (C5b-9). PNH RBCs lack the proteins that normally prevent this cascade from proceeding.
Clinical Features
Hematological
- Anemia - variable, usually mild to moderate; normocytic (may become hypochromic/microcytic with iron deficiency from urinary iron loss)
- Hemoglobinuria - dark "cola-colored" urine, classically in the first morning void
- Hemosiderinuria - almost universally present (chronic urinary iron loss)
- Reticulocytosis - often less than expected for the degree of anemia
- Neutropenia in ~60% of patients
- Thrombocytopenia in ~66% of patients
- Pancytopenia is common
Thrombosis - The Leading Cause of Death
- ~40% of patients develop venous thrombosis
- Veins involved in 85% of cases; often at unusual sites:
- Hepatic veins (Budd-Chiari syndrome)
- Cerebral veins
- Portal and mesenteric veins
- Splenic, renal veins
- Mechanisms: NO scavenging by free hemoglobin causes vasoconstriction/platelet activation; CD59 deficiency on platelets causes phosphatidylserine externalization, forming a scaffold for prothrombinase complexes; endothelial damage from MAC
Abdominal Pain
- ~1/3 of patients; related to smooth muscle dystonias from free hemoglobin scavenging nitric oxide (NO), causing esophageal spasm and intestinal dysmotility
Other Features
- Erectile dysfunction (NO depletion)
- Dysphagia, odynophagia
- Fatigue (profound, even out of proportion to anemia - from NO depletion)
- Pulmonary hypertension (chronic)
- Renal impairment (from hemosiderin deposition and microthrombosis)
- ~5% progress to AML or myelodysplastic neoplasm
Association with Aplastic Anemia and MDS
- PNH clones are present in 50-60% of aplastic anemia (AA) patients
- Present in 15-20% of MDS patients
- Hypothesis: In AA (an autoimmune disease), the immune attack targets GPI-linked antigens on normal HSCs. PIGA-mutant cells lack these GPI antigens and therefore escape immune destruction, gaining a clonal selective advantage.
- PNH and AA thus represent different ends of the same disease spectrum
PNH Clinical Categories
- Classic PNH - hemolysis dominant, large clone, little or no cytopenias from marrow failure
- PNH in the setting of another bone marrow disorder (AA, MDS) - cytopenias predominant
- Subclinical PNH - small clone detected by flow cytometry, no clinical hemolysis
Diagnosis
Flow Cytometry (Gold Standard)
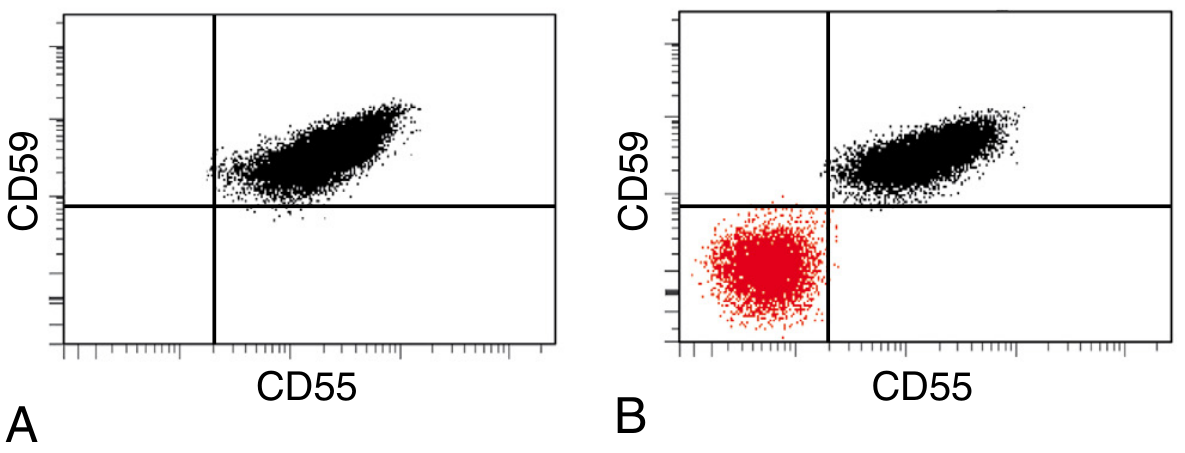
- Detects GPI-deficient cells using antibodies against CD55, CD59 on RBCs
- For granulocytes: CD24 (neutrophils), CD14 (monocytes) are excellent targets
- FLAER (Fluorescein-labeled aerolysin) - binds directly to the GPI anchor; increasingly used for high-sensitivity detection, especially in granulocytes/monocytes
- Guidelines for high-sensitivity flow cytometry to detect minor clones are now widely adopted
Old/Obsolete Tests (Largely Replaced by Flow Cytometry)
- Ham's test (Acid hemolysis test) - lysis of PNH RBCs in acidified serum; classic but insensitive
- Sucrose hemolysis test (Sugar water test) - lysis in low-ionic-strength sucrose solution; screening test, not specific
Other Laboratory Findings
| Test | Finding in PNH |
|---|---|
| LDH | Markedly elevated (marker of intravascular hemolysis) |
| Serum haptoglobin | Low/absent |
| Free plasma hemoglobin | Elevated |
| Urine hemosiderin | Positive (almost always) |
| Urine hemoglobin | Positive during attacks |
| Direct Coombs test | Negative (complement-mediated, not antibody) |
| Reticulocyte count | Elevated but less than expected |
| Bone marrow | Hypercellular (erythroid hyperplasia) or hypocellular |
| PIGA gene sequencing | >120 mutations identified; confirms diagnosis |
Treatment
Complement Inhibitors (Disease-Modifying Therapy)
| Drug | Target | Route | Notes |
|---|---|---|---|
| Eculizumab | C5 (prevents MAC) | IV every 2 weeks | First approved (2007); reduces IVH and thrombosis by up to 90% |
| Ravulizumab | C5 (long-acting) | IV every 8 weeks | Same mechanism, less frequent dosing |
| Pegcetacoplan | C3 (upstream) | SC twice weekly | Prevents both intravascular AND extravascular hemolysis; corrects anemia more completely |
| Danicopan | Factor D (alternative pathway) | Oral | Add-on for extravascular hemolysis on C5 inhibitors |
| Iptacoplan | Factor B (alternative pathway) | Oral | Upstream blockade |
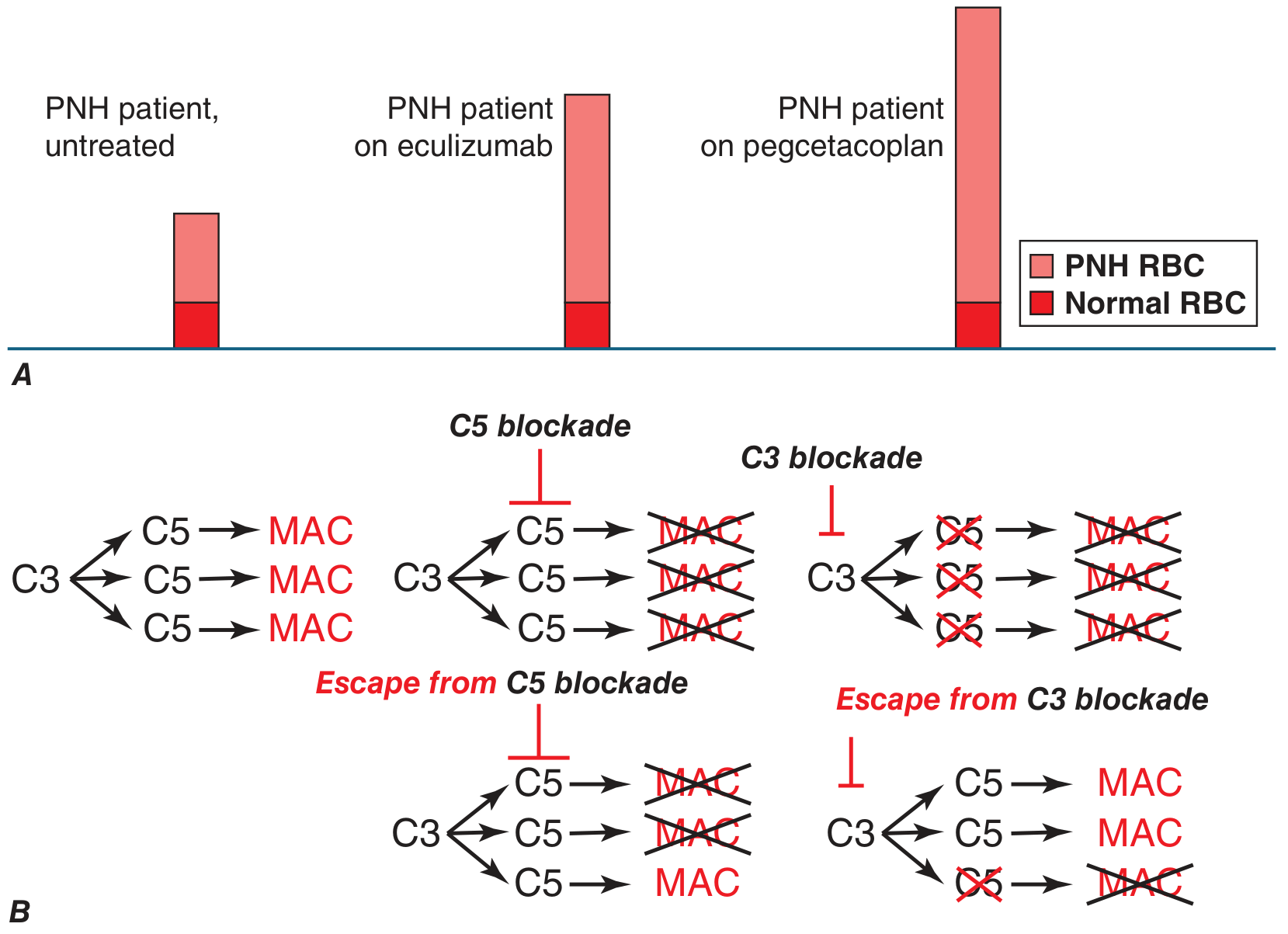
Key distinction - eculizumab vs. pegcetacoplan:
- On eculizumab: MAC formation is prevented, so PNH RBCs survive - but they still bind C3 fragments (opsonization), causing extravascular hemolysis by macrophages. Coombs test becomes positive. Mild residual anemia persists.
- On pegcetacoplan (C3 blockade): Both intravascular AND extravascular hemolysis are prevented; anemia is fully corrected.
Critical risk: Meningococcal infection
All patients on complement inhibitors must receive meningococcal vaccination (quadrivalent + serogroup B vaccines) before starting therapy, and revaccinate every 3-5 years, due to high risk of fatal Neisseria meningitidis infection.
Supportive Treatment
- Iron and folate supplementation (compensate for urinary losses and increased erythropoiesis)
- Red cell transfusions (washed RBCs preferred)
- Anticoagulation for thrombosis (heparin acutely; warfarin or LMWH long-term)
- Eculizumab/anticoagulation significantly reduces, but does not eliminate, thrombotic risk
Definitive Cure
- Allogeneic hematopoietic stem cell transplantation (HSCT) - the only cure; typically reserved for patients with intercurrent aplastic anemia, myeloid malignancy, or refractory disease
Summary
| Feature | Key Point |
|---|---|
| Cause | Somatic PIGA mutation in HSC → no GPI anchors → loss of CD55/CD59 |
| Hemolysis type | Intravascular (complement-mediated, MAC) |
| DAT (Coombs) | Negative (unless on eculizumab) |
| Thrombosis | Major cause of death; unusual sites (hepatic, cerebral veins) |
| Diagnosis | Flow cytometry (CD55/CD59 on RBCs; FLAER on granulocytes) |
| Treatment | Complement inhibitors (eculizumab/ravulizumab/pegcetacoplan) |
| Cure | Allogeneic HSCT |
| Association | Aplastic anemia (50-60%), MDS (15-20%) |
Sources: Robbins & Kumar Pathologic Basis of Disease (10th ed.) | Henry's Clinical Diagnosis and Management by Laboratory Methods | Harrison's Principles of Internal Medicine, 22nd ed. (2025) | Robbins, Cotran & Kumar Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.