Mechanism of Anticoagulants (heparin, warfarin), antiplatelet, fibrinolytic drugs
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have all the material needed. Here is the comprehensive answer:
Mechanisms of Anticoagulants, Antiplatelet, and Fibrinolytic Drugs
1. ANTICOAGULANTS
A. Heparin (Unfractionated Heparin - UFH)
Structure: A sulfated polysaccharide derived from porcine intestinal mucosa. It is a polymer of alternating D-glucuronic acid and N-acetyl-D-glucosamine residues, with a mean molecular weight of 15,000 (range 5,000-30,000).
Mechanism of Action:
Heparin works by activating antithrombin (previously called antithrombin III), a serine protease inhibitor (serpin) synthesized in the liver. Antithrombin acts as a "suicide substrate" for its target coagulation enzymes.
The mechanism has two components:
-
Conformational change (Factor Xa inhibition): Heparin binds to antithrombin via a unique pentasaccharide sequence found on about one-third of commercial heparin chains. This binding induces a conformational change in the reactive center loop of antithrombin, making it more accessible to target proteases. This accelerates Factor Xa inhibition by at least two orders of magnitude.
-
Template mechanism (Thrombin inhibition): To inhibit thrombin, heparin must simultaneously bind to both antithrombin AND thrombin, forming a ternary complex. This requires heparin chains of at least 18 saccharide units (MW ≥ 5,400). Since UFH has a mean MW of 15,000, all its chains are long enough for this bridging function - so UFH inhibits both Factor Xa AND thrombin (IIa) equally.
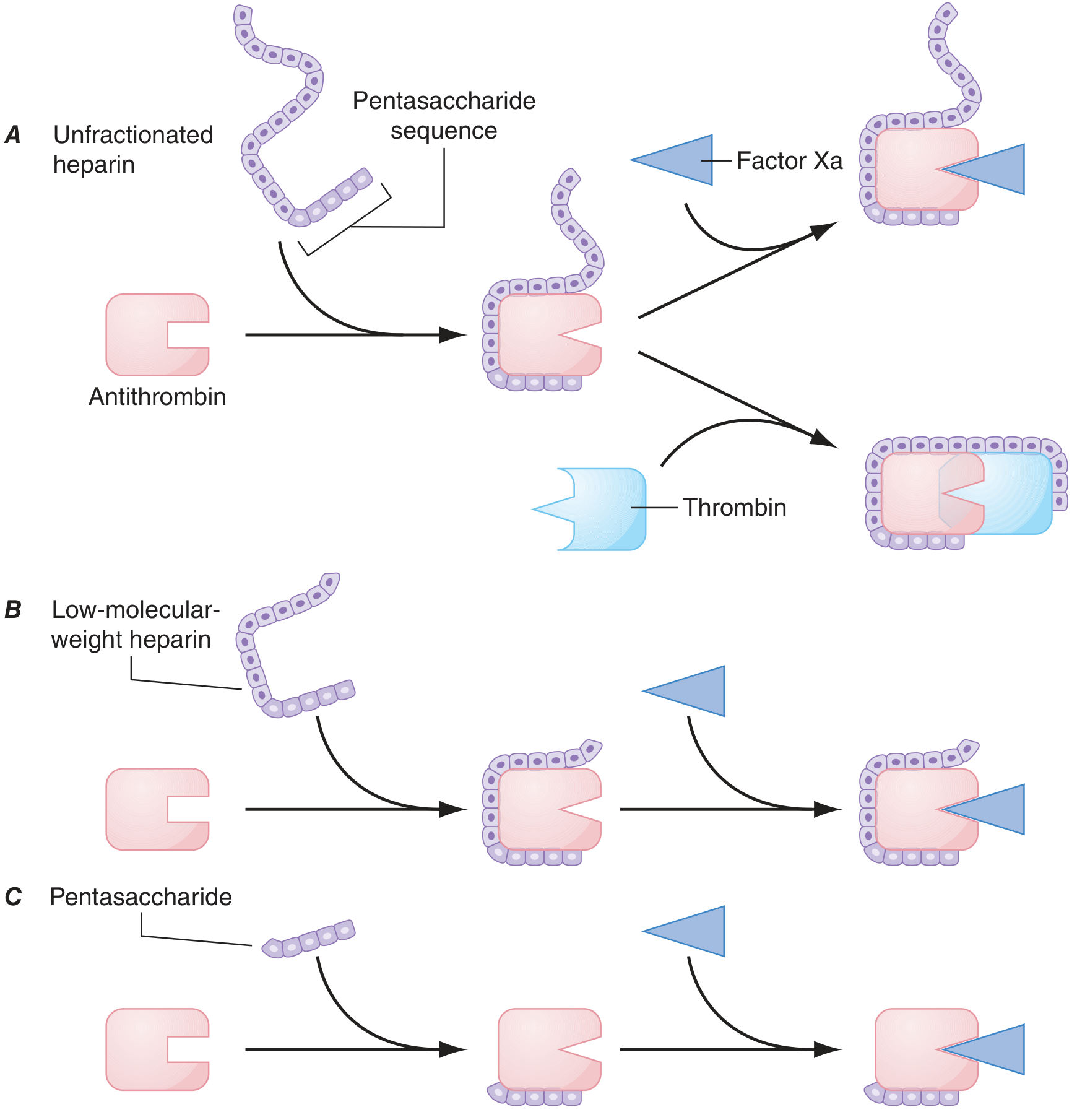
Figure: UFH (A) inhibits both Factor Xa and Thrombin. LMWH (B) preferentially inhibits Factor Xa. Fondaparinux (C) only inhibits Factor Xa.
Net effect: Inhibition of thrombin (IIa), Xa, IXa, XIa, XIIa → prevents clot formation
Monitoring: aPTT (activated partial thromboplastin time); target 60-80 seconds (1.5-2.5x normal)
Reversal: Protamine sulfate (1 mg neutralizes 100 units heparin)
B. Low-Molecular-Weight Heparin (LMWH - e.g., Enoxaparin)
Structure: Mean MW of 4,500-5,000 (about 15-17 saccharide units). Produced by depolymerization of UFH.
Mechanism: Like UFH, it activates antithrombin via the pentasaccharide sequence. However, because at least half of LMWH chains are too short to simultaneously bridge antithrombin to thrombin, LMWH has:
- Greater anti-Xa activity (retained)
- Less anti-IIa (thrombin) activity (reduced)
- Anti-Xa : anti-IIa ratio = 2:1 to 4:1
Advantages over UFH: More predictable dose response, longer half-life (~4 h), less HIT risk, no routine monitoring needed.
Monitoring: Anti-Xa levels (in renal impairment, pregnancy, obesity)
C. Fondaparinux (Synthetic Pentasaccharide)
A synthetically derived pentasaccharide that selectively binds antithrombin, potentiating Factor Xa inhibition by 300- to 1,000-fold. Being only 5 saccharides long, it cannot bridge antithrombin to thrombin. Therefore, it only inhibits Factor Xa, with NO anti-IIa activity. Does not cause HIT (does not bind PF4). Half-life = 17 hours; once-daily SC dosing; renally excreted.
D. Warfarin (Vitamin K Antagonist)
Mechanism of Action:
Warfarin acts by inhibiting Vitamin K Epoxide Reductase (VKOR), blocking the regeneration of the active reduced form of Vitamin K.
The pathway:
- Coagulation factors II, VII, IX, X (plus protein C and S) require gamma-carboxylation of their glutamic acid residues to form γ-carboxyglutamic acid (Gla) residues.
- These Gla residues bind calcium ions, which are essential for the factors to interact with anionic phospholipid surfaces on platelet membranes and activate coagulation.
- The γ-carboxylation reaction requires reduced vitamin K as a cofactor. During the reaction, reduced vitamin K is converted to vitamin K epoxide.
- Vitamin K epoxide must be recycled back to reduced vitamin K by vitamin K epoxide reductase (VKOR).
- Warfarin inhibits VKOR → vitamin K cannot be recycled → insufficient γ-carboxylation → factors II, VII, IX, X are produced with diminished activity (only 10-40% of normal).
Key points:
- Warfarin is a racemic mixture of R and S enantiomers; the S-isomer is ~5x more potent and is metabolized by CYP2C9
- Onset is delayed 72-96 hours (time needed to deplete existing fully-active circulating clotting factors)
- Factor VII has the shortest half-life (~6h), so PT/INR rises first
- The antithrombotic effect depends on depletion of factor X (t½ = 24h) and prothrombin/II (t½ = 72h)
- During initiation, there is a prothrombotic window because protein C (t½ = 8h) and protein S are also depleted faster than the procoagulant factors - hence overlap with heparin for ≥5 days
- Monitoring: INR (target 2.0-3.0 for most indications)
- Reversal: Vitamin K1 (slow), Fresh Frozen Plasma/PCC (fast)
- Genetic factors: CYP2C9 polymorphisms affect S-warfarin metabolism; VKORC1 polymorphisms affect enzyme sensitivity
Harrison's Principles of Internal Medicine, 22E, p. 994; Lippincott Pharmacology, p. 457
2. ANTIPLATELET DRUGS
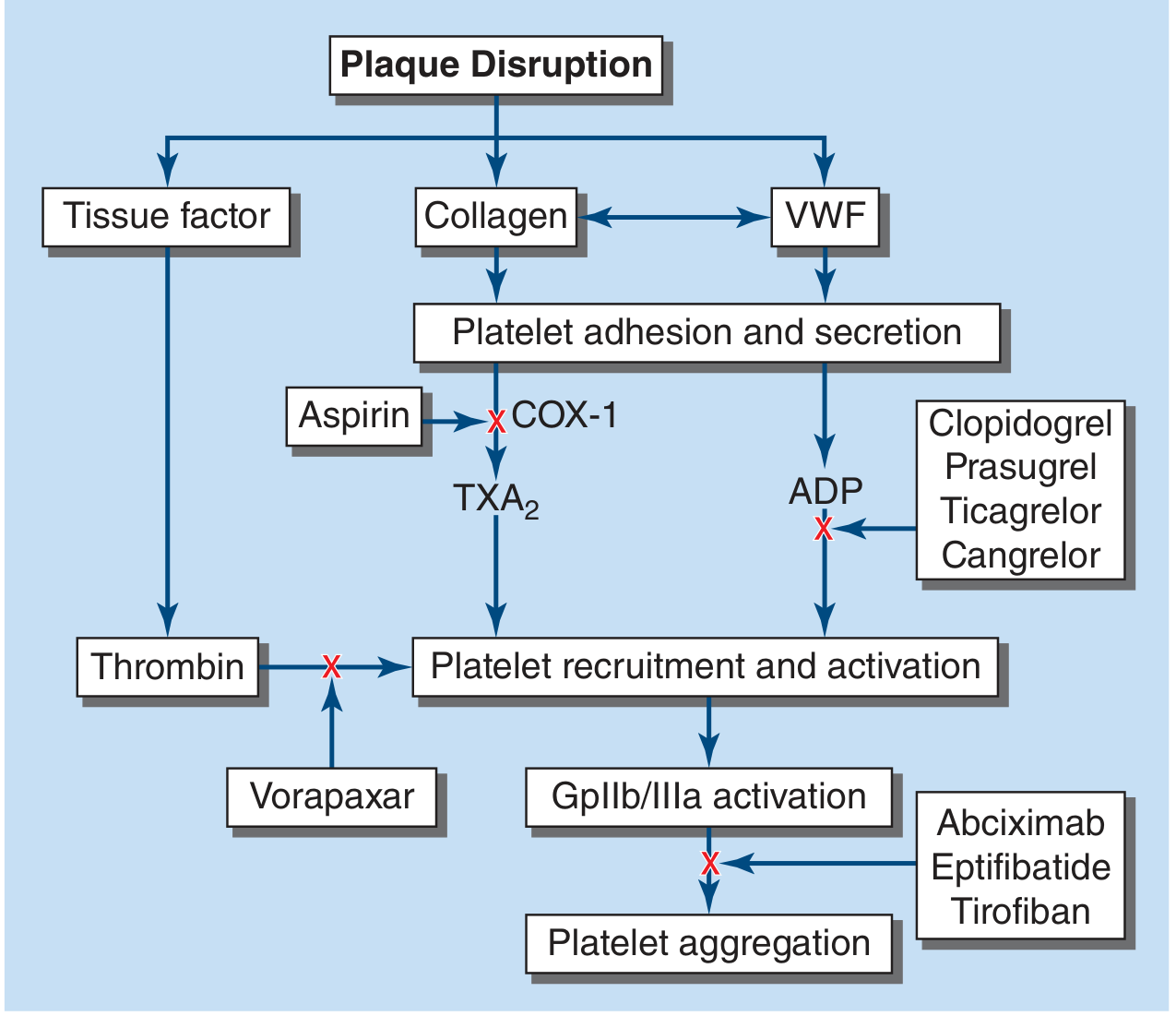
Figure: Sites of action of antiplatelet drugs - from Harrison's Principles of Internal Medicine, 22E
A. Aspirin
Mechanism: Aspirin irreversibly acetylates and inhibits Cyclooxygenase-1 (COX-1) in platelets, blocking the biosynthesis of Thromboxane A2 (TXA2). TXA2 is a potent platelet activator and vasoconstrictor. Since platelets lack a nucleus and cannot synthesize new COX-1, the inhibition lasts for the entire lifespan of the platelet (~7-10 days).
- At high doses (~1 g/day): also inhibits COX-2 in endothelial cells, blocking prostacyclin (PGI2) synthesis - which would counteract the antiplatelet effect.
- Low doses (75-100 mg/day) are preferred to spare prostacyclin production.
Dose: 75-325 mg/day; 160 mg for rapid effect
B. P2Y12 Receptor Antagonists (ADP Receptor Blockers)
| Drug | Reversibility | Prodrug | Notes |
|---|---|---|---|
| Clopidogrel | Irreversible | Yes (CYP2C19) | Most widely used |
| Prasugrel | Irreversible | Yes (CYP3A4/CYP2B6) | Faster, more potent |
| Ticagrelor | Reversible | No | Direct-acting |
| Cangrelor | Reversible | No | IV, very short t½ |
Mechanism: Block the P2Y12 ADP receptor on platelet surfaces, preventing ADP-mediated platelet activation. ADP released from activated platelets normally amplifies platelet aggregation - blocking this receptor interrupts the positive feedback loop.
- Clopidogrel and prasugrel = irreversible P2Y12 blockers (thienopyridines, prodrugs requiring hepatic CYP450 activation)
- Ticagrelor and cangrelor = reversible P2Y12 blockers (direct-acting, no prodrug activation needed)
C. GPIIb/IIIa Inhibitors
Drugs: Abciximab, eptifibatide, tirofiban
Mechanism: Block the final common pathway of platelet aggregation by inhibiting the GPIIb/IIIa receptor (activated integrin αIIbβ3) from binding fibrinogen and von Willebrand factor (vWF). This prevents cross-linking of platelets, stopping aggregation entirely. IV administration only.
D. Vorapaxar
Mechanism: Inhibits PAR-1 (Protease-Activated Receptor-1), the major thrombin receptor on human platelets, thereby blocking thrombin-mediated platelet activation.
Harrison's Principles of Internal Medicine, 22E, p. 986; Goodman & Gilman's Pharmacological Basis of Therapeutics
3. FIBRINOLYTIC (THROMBOLYTIC) DRUGS
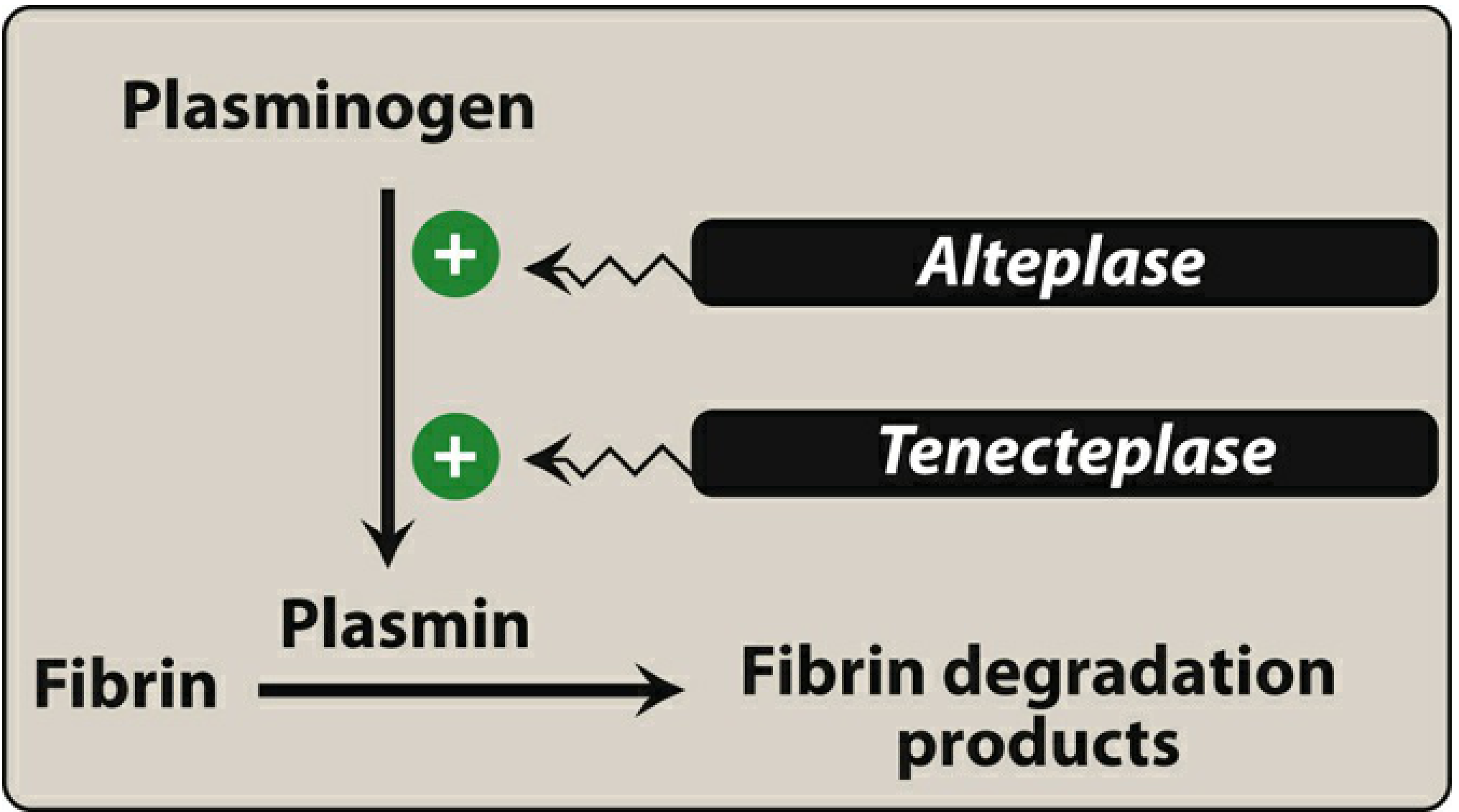
Figure: Activation of plasminogen by thrombolytic drugs (Lippincott Pharmacology)
Core Mechanism (All Fibrinolytics)
All fibrinolytic agents act by activating plasminogen to plasmin (directly or indirectly). Plasmin is a serine protease that cleaves fibrin, dissolving the thrombus. Fibrin degradation products (FDPs, including D-dimers) are released.
Key principle: Clots become more resistant to lysis over time, so earlier treatment yields better results.
Individual Agents
| Agent | Type | Fibrin Selectivity | Notes |
|---|---|---|---|
| Streptokinase | Indirect activator | Non-selective | Bacterial protein; forms complex with plasminogen, antigenic |
| Urokinase | Direct activator | Non-selective | Derived from urine/renal cells |
| Alteplase (tPA) | Direct activator | Fibrin-selective | Low affinity for free plasminogen; activated ~1,000x by fibrin-bound plasminogen |
| Reteplase (rPA) | Direct activator | Fibrin-selective | Longer t½ than alteplase; double bolus IV |
| Tenecteplase (TNK) | Direct activator | Fibrin-selective | Longest t½; single IV bolus; highest fibrin selectivity |
Streptokinase mechanism: An indirect activator - forms a 1:1 complex with plasminogen, which then undergoes a conformational change and acts enzymatically to convert other plasminogen molecules to plasmin. Non-fibrin selective; systemic fibrinolytic state.
tPA (Alteplase) mechanism: Has low affinity for free plasminogen in circulation, but rapidly activates plasminogen bound to fibrin within a thrombus (fibrin acts as a cofactor, enhancing tPA activity ~1,000-fold). This makes it relatively "fibrin-selective" - targeting the clot rather than circulating plasminogen.
Major adverse effect: Bleeding (cannot distinguish pathological thrombus from beneficial hemostatic plug). Contraindicated in: recent surgery/trauma, intracranial pathology, active internal bleeding, uncontrolled hypertension.
Antifibrinolytics (reversal): Tranexamic acid and aminocaproic acid - block the lysine-binding sites on plasminogen, preventing its binding to fibrin.
Lippincott Pharmacology, pp. 464-466; Harrison's Principles of Internal Medicine, 22E
Quick Summary Table
| Drug Class | Example | Target | Mechanism |
|---|---|---|---|
| UFH | Heparin | AT → IIa + Xa | Binds AT, conformational change + bridging |
| LMWH | Enoxaparin | AT → Xa > IIa | Binds AT, conformational change only (mostly) |
| Pentasaccharide | Fondaparinux | AT → Xa only | Binds AT, only Xa inhibition |
| Vitamin K antagonist | Warfarin | Factors II,VII,IX,X | Inhibits VKOR → blocks γ-carboxylation |
| COX-1 inhibitor | Aspirin | TXA2 synthesis | Irreversible COX-1 acetylation |
| P2Y12 blocker | Clopidogrel | ADP receptor | Irreversible P2Y12 blockade |
| GPIIb/IIIa inhibitor | Abciximab | GPIIb/IIIa | Blocks fibrinogen/vWF binding |
| Fibrinolytic | Alteplase | Plasminogen | Activates plasminogen → plasmin → fibrin lysis |
| Fibrinolytic | Streptokinase | Plasminogen | Forms complex with plasminogen → plasmin |
This is a shared conversation. Sign in to Orris to start your own chat.