Write about etiopathogenesis, complications of Dilated Cardiomyopathy
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Searching PubMed
"dilated cardiomyopathy"[MeSH Terms] AND pathogenesis
I have all the content I need. Let me now compose the comprehensive answer.
Dilated Cardiomyopathy (DCM): Etiopathogenesis and Complications
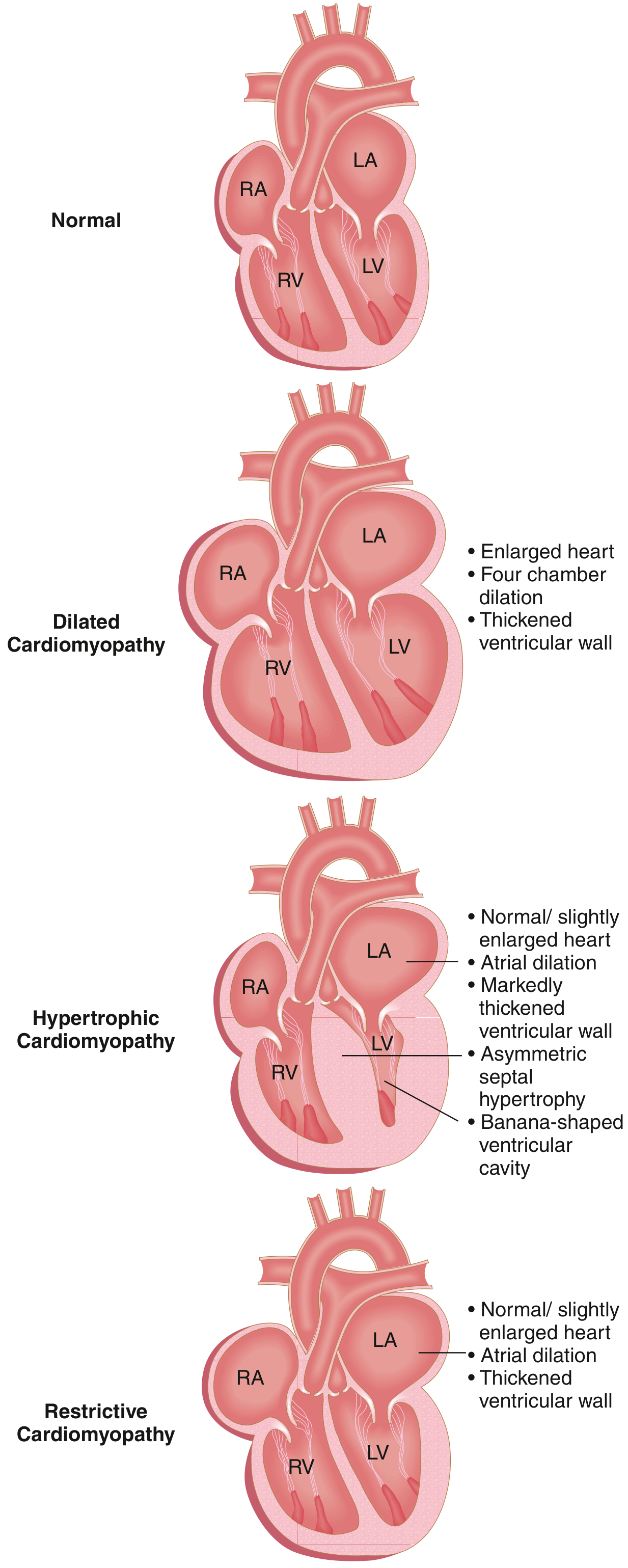
Fig. 9.23 - The three major forms of cardiomyopathy. In DCM, all four chambers dilate, ventricular wall thickness varies (thickened, normal, or thinned), and systolic dysfunction predominates. — Robbins & Kumar Basic Pathology
Definition
Dilated cardiomyopathy (DCM) is a heart muscle disorder defined by progressive cardiac dilation and impaired systolic (contractile) dysfunction of the left ventricle or both ventricles, in the absence of coronary artery disease, valvular abnormalities, or pericardial disease. It is the most common form of cardiomyopathy (90% of cases), with an estimated prevalence of 1 in 250 adults. DCM is most commonly diagnosed between 20 and 50 years of age.
Etiopathogenesis
The damage that culminates in end-stage DCM can be initiated by inherited abnormalities or environmental exposures. At diagnosis, the disease has usually progressed to end-stage heart failure with poor contractility and does not reveal specific pathologic features.
1. Genetic Causes (20-50% of cases)
- Over 50 genes are known to be mutated in DCM; autosomal dominant inheritance is the predominant pattern.
- Mutations primarily affect cytoskeletal proteins or proteins linking the sarcomere to the cytoskeleton (loss-of-function mutations).
- Key mutated genes include:
- TTN (Titin) - the most common; accounts for ~25% of familial cases and ~18% of sporadic cases. Titin spans the sarcomere connecting the Z and M bands, limiting passive range of motion during stretching.
- β-myosin heavy chain (MYH7)
- α-myosin heavy chain (MYH6)
- Cardiac troponin T (TNNT2)
- Desmin - the principal intermediate filament protein in cardiac myocytes
- Lamin A/C - nuclear envelope protein; mutations cause atrial/ventricular arrhythmias and progressive AV conduction disease (which may precede DCM)
- Filamin C (FLNC) - truncating mutations cause arrhythmogenic + dilated phenotype with lethal ventricular arrhythmias in adolescents and heart failure in survivors
- X-linked DCM (2-5% of familial cases): caused by mutations in dystrophin, a membrane protein that physically couples the intracellular cytoskeleton to the extracellular matrix. It is associated with Duchenne, Becker, and Emery-Dreifuss muscular dystrophies (accounting for 26% of cases, 90% of which are these dystrophies).
- Note: Gain-of-function mutations in some of the same sarcomere genes (e.g., MYH7, TNNT2) cause hypertrophic cardiomyopathy instead.
- Since contractile myocytes and conduction fibers share a common developmental pathway, conduction abnormalities can also be a feature of inherited DCM.
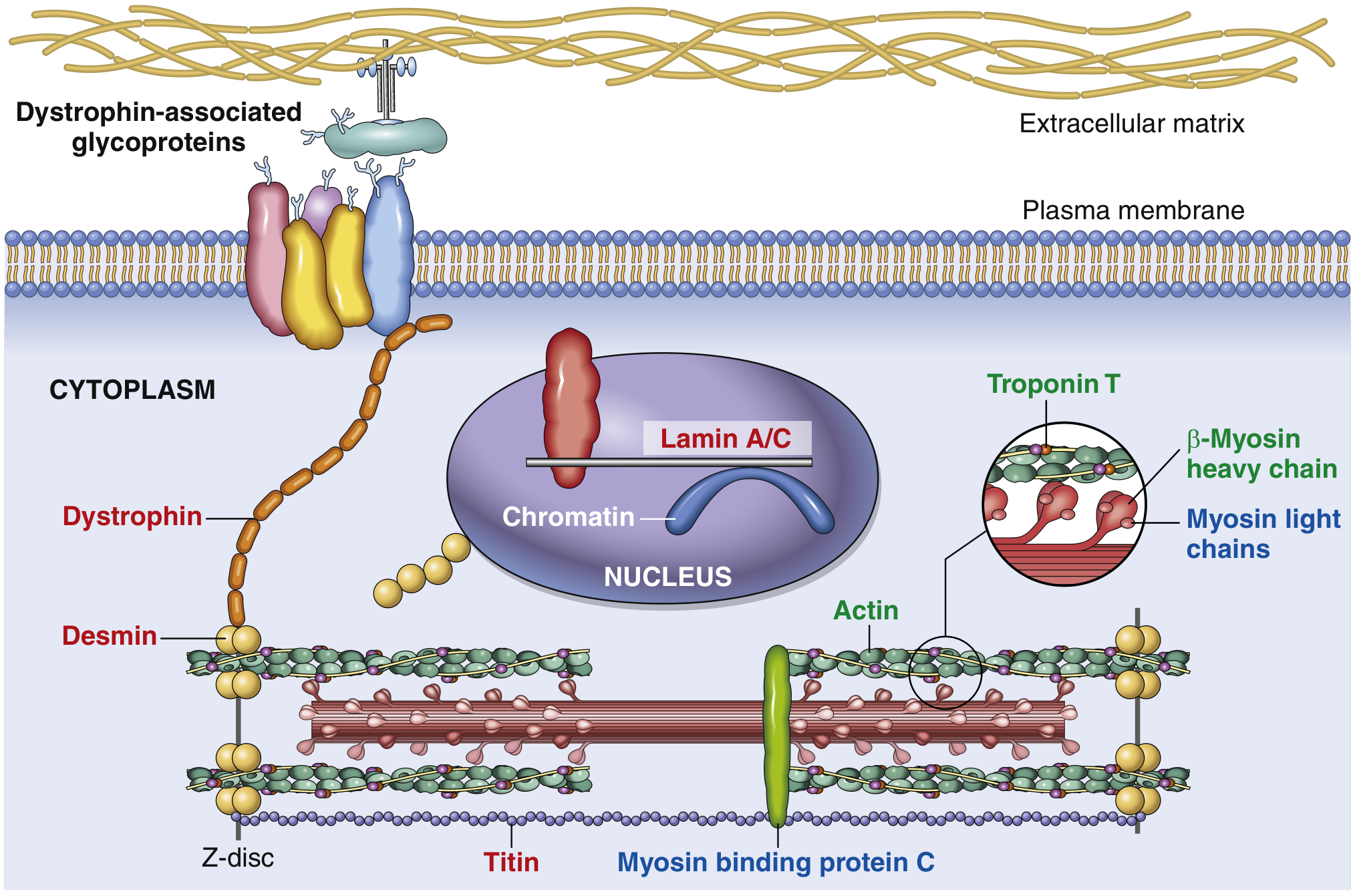
Fig. 9.24 - Schematic of a myocyte showing key proteins mutated in DCM (red labels), HCM (blue labels), or both (green labels). Titin, the largest known human protein (~30,000 amino acids), accounts for ~20% of all DCM. — Robbins & Kumar Basic Pathology
2. Infectious / Viral Myocarditis
- Older implicated organisms: Adenovirus, Enterovirus (coxsackievirus B), echovirus, Epstein-Barr virus, CMV
- More recently identified: Parvovirus B-19 and human herpesvirus 6 (HHV-6)
- Mechanism: Nucleic acid footprints of coxsackievirus B and enteroviruses can be detected in late-stage DCM myocardium. Sequential endomyocardial biopsies have documented instances where infectious myocarditis progressed to DCM.
- In many cases, viral transcripts or elevated antiviral antibodies may be the only evidence of a "missed" myocarditis, even when inflammation is absent from the end-stage heart.
- Other organisms: Rickettsia, parasites (Chagas disease - Trypanosoma cruzi), fungi
3. Alcohol and Toxic Exposures
- Alcohol: Strongly associated with DCM. Alcohol and its metabolite acetaldehyde have direct toxic effects on the myocardium. Chronic alcohol use disorder can also be associated with nutritional deficiencies (e.g., thiamine - contributing to beriberi heart disease) and autonomic dysfunction.
- Cardiotoxic drugs:
- Anthracyclines (doxorubicin, daunorubicin): Dose-dependent cardiotoxicity
- Cyclophosphamide
- Tyrosine kinase inhibitors and certain immunotherapies
- Radiation therapy: Can cause cardiac fibrosis and secondary DCM.
4. Peripartum (Pregnancy-associated) DCM
- Develops in the peripartum period (last month of pregnancy to 5 months postpartum)
- Mechanism: incompletely understood; proposed roles for prolactin cleavage products (16-kDa prolactin), anti-cardiac antibodies, nutritional deficiencies, and increased hemodynamic demands
- May partially or fully reverse with treatment; may recur in subsequent pregnancies
5. Nutritional Deficiencies
- Thiamine (Vitamin B1) deficiency - wet beriberi
- Selenium deficiency - Keshan disease
- Carnitine deficiency - impairs mitochondrial fatty acid oxidation
6. Iron Overload (Hemochromatosis)
- Marked accumulation of intramyocardial hemosiderin demonstrable by Prussian blue stain
- Iron deposits cause direct myocardial toxicity and cardiomyocyte death
7. Inflammatory and Autoimmune Disorders
- Systemic lupus erythematosus (SLE), systemic sclerosis (scleroderma)
- Sarcoidosis - granulomatous infiltration of the myocardium
- Giant cell myocarditis
8. Endocrinopathies
- Hypothyroidism, hyperthyroidism, acromegaly, pheochromocytoma (catecholamine excess), diabetes mellitus
9. Metabolic Disorders
- Mitochondrial disorders
- Glycogen storage diseases (e.g., Pompe disease)
- Fatty acid oxidation defects
- Mucopolysaccharidoses
10. Tachycardia-mediated Cardiomyopathy (Tachycardiomyopathy)
- A reversible form; cardiomyopathy usually reverses once the tachycardia is controlled.
11. Idiopathic DCM
- When no identifiable cause is found after complete workup, the disease is termed idiopathic DCM, which remains the most common label. Many idiopathic cases may have an unidentified genetic origin.
Morphology (Gross and Histology)
Gross Pathology:
- Heart is characteristically enlarged (up to 2-3 times normal weight) and flabby
- Dilation of all four chambers (biventricular > left-sided predominance)
- Ventricular wall thickness may be less than, equal to, or greater than normal (due to concurrent hypertrophy)
- Mural thrombi are often present (source of thromboemboli)
- Valvular and vascular lesions are absent (by definition)
Histology (nonspecific):
- Most myocytes show hypertrophy with enlarged nuclei
- Many are attenuated, stretched, and irregular
- Variable interstitial and endocardial fibrosis
- Scattered areas of replacement fibrosis (marking previous ischemic necrosis or missed myocarditis)
- In iron overload DCM: hemosiderin deposits (Prussian blue positive)
Complications of Dilated Cardiomyopathy
1. Progressive Congestive Heart Failure (CHF)
The fundamental defect is ineffective contraction. Ejection fraction is typically <25% at end-stage (normal: 50-65%). This leads to:
- Left heart failure: dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, pulmonary edema
- Right heart failure: elevated JVP, hepatomegaly, ascites, peripheral edema
- Low cardiac output: sinus tachycardia, weak pulses, hypotension
- Functional mitral and tricuspid regurgitation due to annular dilation (pansystolic murmur at apex)
- Third (and sometimes fourth) heart sound (S3 gallop)
2. Arrhythmias
- Supraventricular arrhythmias: Atrial fibrillation (most common; increases thromboembolic risk)
- Ventricular arrhythmias: Ventricular tachycardia (VT), ventricular fibrillation (VF) - can cause sudden cardiac death
- Conduction disturbances: AV block, bundle branch block, complete heart block (especially with LMNA or desmin mutations)
- Arrhythmias are particularly common in patients with mutations in TTN, DSP, RBM20, LMNA, and FLNC genes
3. Sudden Cardiac Death (SCD)
- A major cause of death in DCM (along with progressive cardiac failure)
- Caused by ventricular arrhythmias (VF/VT)
- First presentation of DCM may be sudden death
- ICD implantation is used for primary prevention in high-risk patients
4. Thromboembolism
- Mural thrombi form in dilated, poorly contracting ventricles (especially the left ventricular apex) and atria
- Can produce:
- Systemic arterial emboli (stroke, limb ischemia, mesenteric ischemia)
- Pulmonary emboli (from right heart or deep vein thrombosis)
- Anticoagulation is indicated in patients with AF or echocardiographic evidence of mural thrombus
5. Functional Valvular Regurgitation
- Functional mitral regurgitation: Annular dilatation and papillary muscle displacement cause leaflet malcoaptation
- Functional tricuspid regurgitation: Similar mechanism on the right side
- Worsens hemodynamic status and symptoms; may require surgical correction at the time of transplant
6. Pulmonary Hypertension
- Secondary to chronically elevated left ventricular filling pressures
- Causes reactive pulmonary arterial hypertension
- Can complicate cardiac transplantation candidacy
7. Hepatic Complications (Congestive Hepatopathy)
- Chronic right heart failure leads to hepatic congestion
- Cardiac cirrhosis in severe, chronic cases
- Elevated liver enzymes, bilirubin; eventually hepatic failure ("cardiac hepatopathy")
8. Renal Complications
- Reduced cardiac output leads to prerenal azotemia
- Cardiorenal syndrome (low perfusion + neurohormonal activation)
- End-stage heart failure is commonly associated with concomitant renal failure
9. Cachexia and Skeletal Muscle Wasting
- Chronic low cardiac output, neurohormonal activation, and gut hypoperfusion contribute to cardiac cachexia
- Reduced exercise tolerance, fatigue, anorexia
Summary Table
| Feature | Details |
|---|---|
| Most common genetic cause | TTN (titin) mutations (~25% familial, ~18% sporadic) |
| Inheritance | Autosomal dominant (most common), X-linked (dystrophin) |
| Most common viral agents | Parvovirus B19, HHV-6, Coxsackie B |
| EF in end-stage DCM | <25% (normal 50-65%) |
| Gross morphology | Enlarged, flabby, 4-chamber dilation, mural thrombi |
| Major complications | CHF, SCD (arrhythmia), thromboembolism, functional MR |
| Median survival (untreated) | 4-6 years to transplant or death |
| NYHA IV annual mortality | 30-50% |
Sources:
- Robbins & Kumar Basic Pathology, pp. 370-372 (Pathogenesis and Morphology of DCM)
- Goldman-Cecil Medicine, International Edition, pp. 514-516 (Pathobiology, Clinical Manifestations, Treatment)
- Tintinalli's Emergency Medicine: A Comprehensive Study, pp. 875-876 (DCM in children and adults)
Pathogenesis
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive, deep content from Braunwald's Heart Disease, Robbins Pathology, and Goldman-Cecil. I have the key pathogenesis diagrams. Let me compose the detailed pathogenesis answer.
Pathogenesis of Dilated Cardiomyopathy
The pathogenesis of DCM is multifactorial and involves three broad converging pathways: genetic/molecular disruption of sarcomere-cytoskeletal integrity, viral and immune-mediated myocardial injury, and final common pathway of ventricular remodeling and neurohormonal activation. At diagnosis, the disease has usually progressed to end-stage disease with poor contractility and no specific pathologic features that reveal the original cause.
I. Genetic Pathogenesis: Sarcomere-Cytoskeletal Dysfunction
Concept: "The DCM Gene Ontology"
Unlike HCM (caused almost exclusively by sarcomeric protein mutations), the genes implicated in DCM are diverse in subcellular location - spanning the sarcomere, Z-disc, cytoskeleton, nuclear membrane, and ion channels. This is a key distinguishing feature. (Braunwald's Heart Disease)
Mechanism of Force Transmission Failure
The central molecular mechanism in genetic DCM is disruption of the force-transmission chain from sarcomere to extracellular matrix:
Sarcomere → Z-disc → Cytoskeleton → Cell membrane (Dystrophin complex) → ECM
Loss-of-function mutations at any point along this chain impair the myocyte's ability to generate or transmit contractile force, ultimately leading to progressive dilation and systolic dysfunction.
Key Mutated Proteins and Their Roles
| Gene | Protein | Subcellular Location | % of DCM | Mechanism |
|---|---|---|---|---|
| TTN | Titin | Sarcomere (Z-M band scaffold) | 10-25% familial, 10-15% sporadic | Truncating mutations impair sarcomere assembly, passive force generation, and mechanosensing |
| LMNA | Lamin A/C | Inner nuclear membrane | ~6% | Disrupts nuclear structural integrity and gene expression; causes arrhythmia + conduction disease BEFORE DCM |
| MYH7 | β-Myosin heavy chain | Sarcomere (thick filament) | ~4% | Loss-of-function → reduced contractile force (contrast: gain-of-function → HCM) |
| TNNT2 | Cardiac troponin T | Sarcomere (thin filament) | ~3% | Allelic variants cause DCM or HCM depending on the specific mutation |
| DES | Desmin | Intermediate filament | <3% | Links Z-discs to cell membrane and nuclear envelope; loss destabilizes myocyte structure |
| DMD | Dystrophin | Sarcolemma (costamere) | X-linked | Physically couples cytoskeleton to ECM; absence or cleavage leads to membrane disruption |
| FLNC | Filamin C | Z-disc | <3% | Truncating mutations → arrhythmogenic + dilated phenotype with lethal VT in young adults |
| SCN5A | Sodium channel Nav1.5 | Sarcolemma | <2% | Ion channel dysfunction + conduction disease |
Titin (TTN): The Most Important DCM Gene
Titin is the largest known human protein (~30,000-35,000 amino acids), encoded by the TTN gene. It:
- Spans the entire sarcomere, connecting the Z-disc to the M-band
- Acts as a molecular spring (I-band region) and mechanosensor
- Scaffolds sarcomere assembly
- Truncating variants (nonsense, frameshift, splice-site) in TTN impair sarcomere stiffness and elasticity, reducing the passive force that normally keeps the sarcomere in an optimal length-tension relationship
Penetrance of TTN mutations is markedly reduced in individuals of African ancestry compared to European ancestry.
Lamin A/C (LMNA): Arrhythmia-First DCM
LMNA mutations cause a distinctive clinical phenotype: conduction system disease and arrhythmias (AF, AV block, VT) precede the development of DCM. Mutant lamins also cause a spectrum of "laminopathies" including Emery-Dreifuss muscular dystrophy, limb-girdle MD type 1B, lipodystrophy syndromes, and Hutchinson-Gilford progeria - all caused by allelic variants in the same gene.
Genetics of Penetrance and Variable Expressivity
- Familial DCM has age-dependent penetrance: disease manifests in the 4th-7th decade typically, though pediatric onset occurs
- Incomplete penetrance: a carrier of a disease-causing allele may never manifest phenotype
- Variable expressivity: even within the same family with the same rare variant, clinical features differ markedly
- These phenomena are explained by environmental "hits" (hypertension, toxins, viral infections), additional rare variants in other DCM genes, and epigenetic differences
II. Viral and Immune-Mediated Pathogenesis
This is a three-stage process (model derived from animal studies with coxsackievirus B):
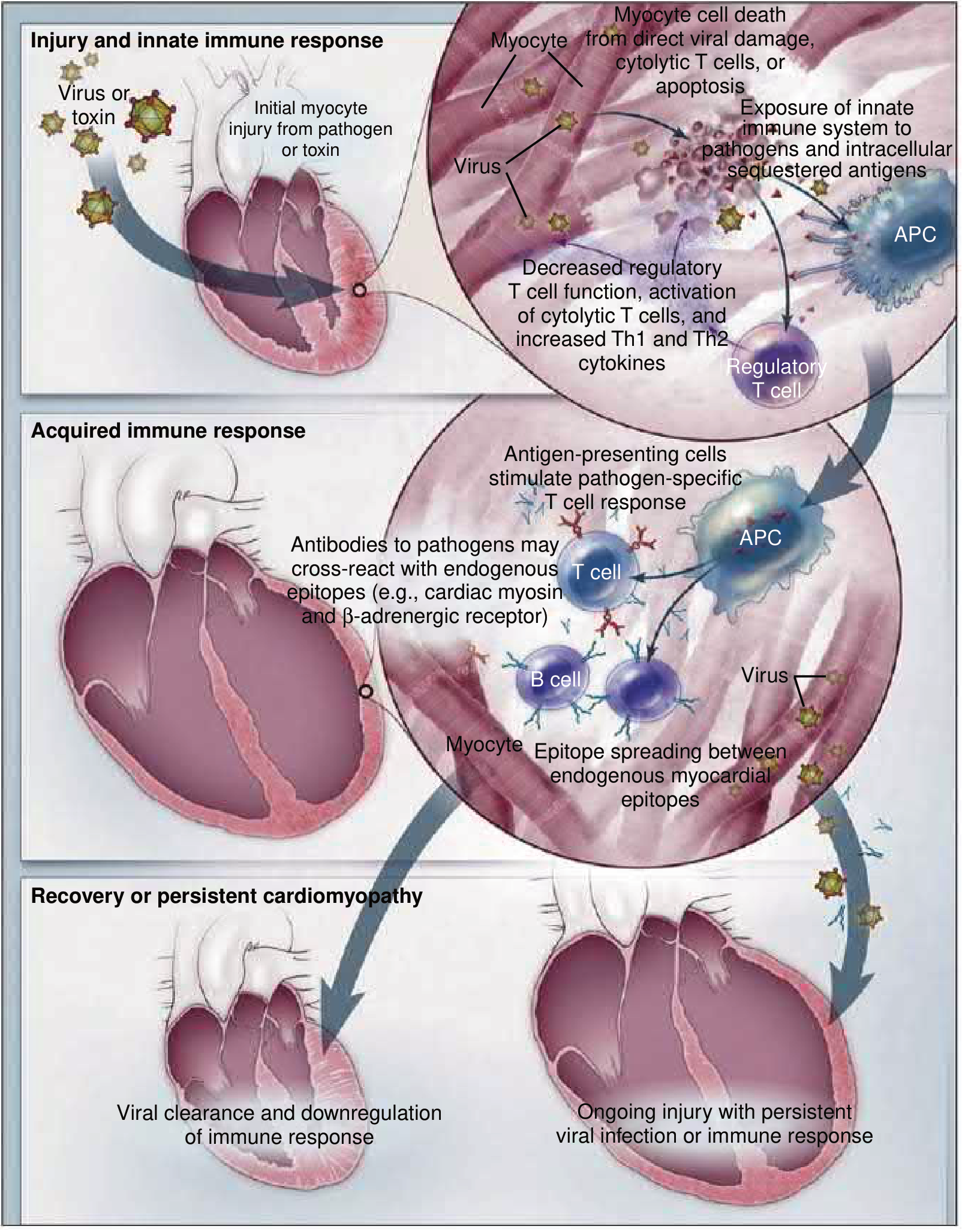
Fig. 55.4 - Pathogenesis of myocarditis progressing to DCM. Three-stage model showing viral injury → innate/acquired immune activation → either viral clearance with recovery, OR persistent infection/immune response leading to chronic DCM. — Braunwald's Heart Disease
Stage 1: Acute Injury and Innate Immune Activation
Viral entry mechanism (e.g., Coxsackievirus B):
- Virus binds the Coxsackievirus-Adenovirus Receptor (CAR) on the myocyte membrane, using DAF (decay-accelerating factor) as a co-receptor
- Receptor engagement activates tyrosine kinases (p56lck, Fyn, Abi) that remodel the host cytoskeleton to facilitate viral entry
- CVB produces protease 2A which cleaves dystrophin - this disrupts the dystrophin-sarcoglycan complex, leading to myocyte membrane damage and facilitates viral release to infect adjacent cells. When dystrophin is absent (Duchenne MD), CVB is released even more efficiently
- Protease 2A and 3C also cleave proteins involved in membrane integrity, translation initiation, apoptosis regulation, and innate immunity
- Viral engagement activates Toll-like receptors (TLRs) via adaptors MyD88 and TRIF
- TLR activation → NF-κB translocation → cytokine production → triggers acquired immunity (CD4+/CD8+ T cell mobilization)
- Alternatively, TLR activation → IRF3 activation → Type I interferon (IFN-β) production, which is protective by attenuating viral replication
Result of Stage 1: Direct myocyte death from viral replication + cytolytic T cells + apoptosis; exposure of intracellular sequestered antigens to the immune system.
Stage 2: Acquired Immune Response
- Antigen-presenting cells (APCs) take up viral and myocardial antigens
- APCs stimulate pathogen-specific T cell responses (CD4+ Th1/Th2 and CD8+ cytotoxic T cells)
- T cells drive B cells to produce anti-viral antibodies
- Key mechanism of injury: molecular mimicry / cross-reactivity - antibodies directed at viral epitopes cross-react with endogenous cardiac antigens:
- Anti-cardiac myosin antibodies
- Anti-β1-adrenergic receptor antibodies
- Anti-mitochondrial antibodies
- Epitope spreading occurs: initial immune response to one cardiac antigen broadens to include other endogenous myocardial epitopes, sustaining inflammation beyond viral clearance
- Decreased regulatory T cell function → loss of immune tolerance → amplified cytotoxic T cell activation
Stage 3: Resolution or Persistent Cardiomyopathy
Outcome A - Recovery: In most patients, the pathogen is cleared and the immune response is downregulated, with few sequelae.
Outcome B - Persistent DCM: In a subset of patients:
- The virus is not fully cleared; persistent viral genomes (even without active replication) sustain immune activation
- Heart-specific autoimmune inflammation persists due to mistaken recognition of endogenous cardiac antigens as foreign (molecular mimicry)
- Chronic myocyte loss, fibrosis, and progressive dilation → end-stage DCM
This explains why DCM is diagnosed months to years after an often-unrecognized viral myocarditis.
III. Alcohol and Toxic Pathogenesis
- Acetaldehyde (alcohol metabolite) has direct cardiotoxic effects: impairs mitochondrial function, increases reactive oxygen species (ROS), disrupts calcium homeostasis, and inhibits myofibrillar protein synthesis
- Chronic alcohol also causes thiamine deficiency, autonomic dysfunction, and electrolyte disturbances
- Anthracyclines (doxorubicin): generate free radicals via redox cycling with Fe²⁺, causing oxidative stress, mitochondrial DNA damage, and myocyte apoptosis. Dose-dependent and largely irreversible.
- Other toxins: cobalt, cocaine (coronary spasm + direct toxicity), methamphetamine
IV. Final Common Pathway: Ventricular Remodeling and Neurohormonal Activation
Regardless of the initiating cause, all forms of DCM converge on a final common pathophysiological pathway:
Initial myocardial injury / cardiomyocyte loss
↓
Reduced Cardiac Output + Increased Wall Stress
↓
Neurohormonal Activation:
• Sympathetic nervous system (↑catecholamines)
• RAAS (↑angiotensin II, ↑aldosterone)
• ADH (↑arginine vasopressin)
↓
Short-term: compensatory (↑HR, ↑contractility, ↑Na+/H₂O retention)
Long-term: maladaptive
↓
Ventricular Remodeling:
• Cardiomyocyte hypertrophy
• Myocyte elongation ("slippage")
• Interstitial fibrosis (TGF-β, angiotensin II-mediated)
• Apoptosis + necrosis of myocytes
• Cytokine overexpression (TNF-α, IL-1, IL-6)
• Vascular and endothelial dysfunction
↓
Progressive cardiac dilation, sphericalization, functional MR
↓
Further reduction in EF → end-stage heart failure
Key mediators in this cascade:
- Angiotensin II: promotes myocyte hypertrophy, fibroblast proliferation, and collagen deposition (interstitial fibrosis)
- Catecholamines: initial positive inotropic effect, but chronic exposure causes β1-receptor downregulation, direct myocyte toxicity, and pro-arrhythmic remodeling
- TNF-α, IL-1β, IL-6: cause myocyte apoptosis, depress contractility, and promote fibrosis
- TGF-β: key driver of myocardial fibrosis (replacement and reactive)
- Aldosterone: promotes sodium retention, fibrosis, and ventricular remodeling
- Nitric oxide (NO) dysregulation: in DCM, NOS3 upregulation in a stressed myocardium can impair contractility
V. Peripartum DCM: Special Pathogenesis
An additional proposed mechanism unique to peripartum DCM:
- Cathepsin D cleaves prolactin into a 16-kDa fragment that is cardiotoxic (causes coronary microvascular endothelial apoptosis, impairs angiogenesis)
- This explains the potential therapeutic role of bromocriptine (prolactin inhibitor) in peripartum DCM
Summary Diagram of Pathogenesis
GENETIC DEFECTS VIRAL/IMMUNE TOXINS/ACQUIRED
(TTN, LMNA, MYH7...) (CVB → myocarditis (Alcohol, anthracyclines,
Sarcomere/cytoskeletal → autoimmunity) peripartum, iron overload)
dysfunction ↓
↘ ↙ ↘ ↙
MYOCYTE LOSS / DYSFUNCTION
↓
NEUROHORMONAL ACTIVATION
(SNS ↑, RAAS ↑, cytokines ↑)
↓
VENTRICULAR REMODELING
(dilation, fibrosis, apoptosis)
↓
DILATED CARDIOMYOPATHY (EF <40%, 4-chamber dilation)
Sources:
- Braunwald's Heart Disease, 2-Vol Set, Chapters 52 and 55 (Genetics and Myocarditis pathogenesis)
- Robbins & Kumar Basic Pathology, pp. 370-372
- Goldman-Cecil Medicine, International Edition, pp. 514-515
- Schwartz's Principles of Surgery, 11e, p. 859
50 years male sudden death, autopsy of the heart revealed gray white area in left ventricle. What is the diagnosis? What are the morphological changes in heart during autopsy to identify the age of lesion?
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive answer. Let me compose it.
Diagnosis and Morphological Age-Dating of Myocardial Infarction at Autopsy
Diagnosis
Myocardial Infarction (Old / Healed) - Fibrous Scar
A gray-white area in the left ventricle at autopsy in a 50-year-old male who died suddenly represents a healed myocardial infarct - a fibrous scar replacing previously infarcted myocardium. The gray-white, hard, shrunken appearance indicates the lesion is at least 6 weeks to >2 months old, where dead muscle has been completely replaced by dense collagenous scar tissue.
The underlying cause is most commonly atherosclerotic coronary artery disease with prior occlusion of a coronary artery. The sudden death in this case was most likely due to a fatal ventricular arrhythmia (VF/VT) triggered by the electrically unstable scar tissue, or a new acute ischemic event superimposed on pre-existing disease.
"By the end of 6 weeks, the infarcted area is replaced by a thin grey-white, hard, shrunken fibrous scar which is well developed in about three months."
- The Essentials of Forensic Medicine & Toxicology, 36th ed.
Location of Infarcts by Coronary Artery Involved
| Coronary Artery | Frequency | Zone Infarcted |
|---|---|---|
| Left anterior descending (LAD) | 40-50% | Anterior LV wall near apex; anterior interventricular septum; apex |
| Right coronary artery (RCA) | 30-40% | Inferior/posterior LV wall; posterior septum; RV free wall (some) |
| Left circumflex (LCx) | 15-20% | Lateral LV wall (except apex) |
Morphological Changes to Age-Date the Infarct
The gross and histological appearance of a myocardial infarct follows a predictable, stereotypical sequence that allows the forensic pathologist to estimate the age of the lesion.
Complete Table of Morphological Changes (Robbins, Cotran & Kumar)
| Time | Gross Features | Light Microscopy | Electron Microscopy |
|---|---|---|---|
| 0-30 min (Reversible injury) | None | None | Relaxation of myofibrils; glycogen loss; mitochondrial swelling |
| 0.5-4 hr (Irreversible begins) | None | Usually none; variable waviness of fibers at border | Sarcolemmal disruption; mitochondrial amorphous densities |
| 4-12 hr | Dark mottling (occasional) | Early coagulative necrosis; edema; hemorrhage | - |
| 12-24 hr | Dark mottling | Ongoing coagulative necrosis; pyknosis of nuclei; myocyte hypereosinophilia; marginal contraction band necrosis; early neutrophilic infiltrate | - |
| 1-3 days | Mottling with yellow-tan infarct center | Coagulative necrosis with loss of nuclei and striations; brisk neutrophilic infiltrate | - |
| 3-7 days | Hyperemic border; central yellow-tan softening | Beginning disintegration of dead myofibers; dying neutrophils; early phagocytosis by macrophages at border; hyperemic border = early granulation tissue | - |
| 7-10 days | Maximally yellow-tan and soft; depressed red-tan margins | Well-developed phagocytosis of dead cells; granulation tissue at margins | - |
| 10-14 days | Red-gray depressed borders | Well-established granulation tissue with new blood vessels and collagen deposition | - |
| 2-8 weeks | Gray-white scar, progressive from border to core | Increased collagen deposition; decreased cellularity | - |
| >2 months | Scarring complete | Dense collagenous scar | - |
Histological Sequence in Detail
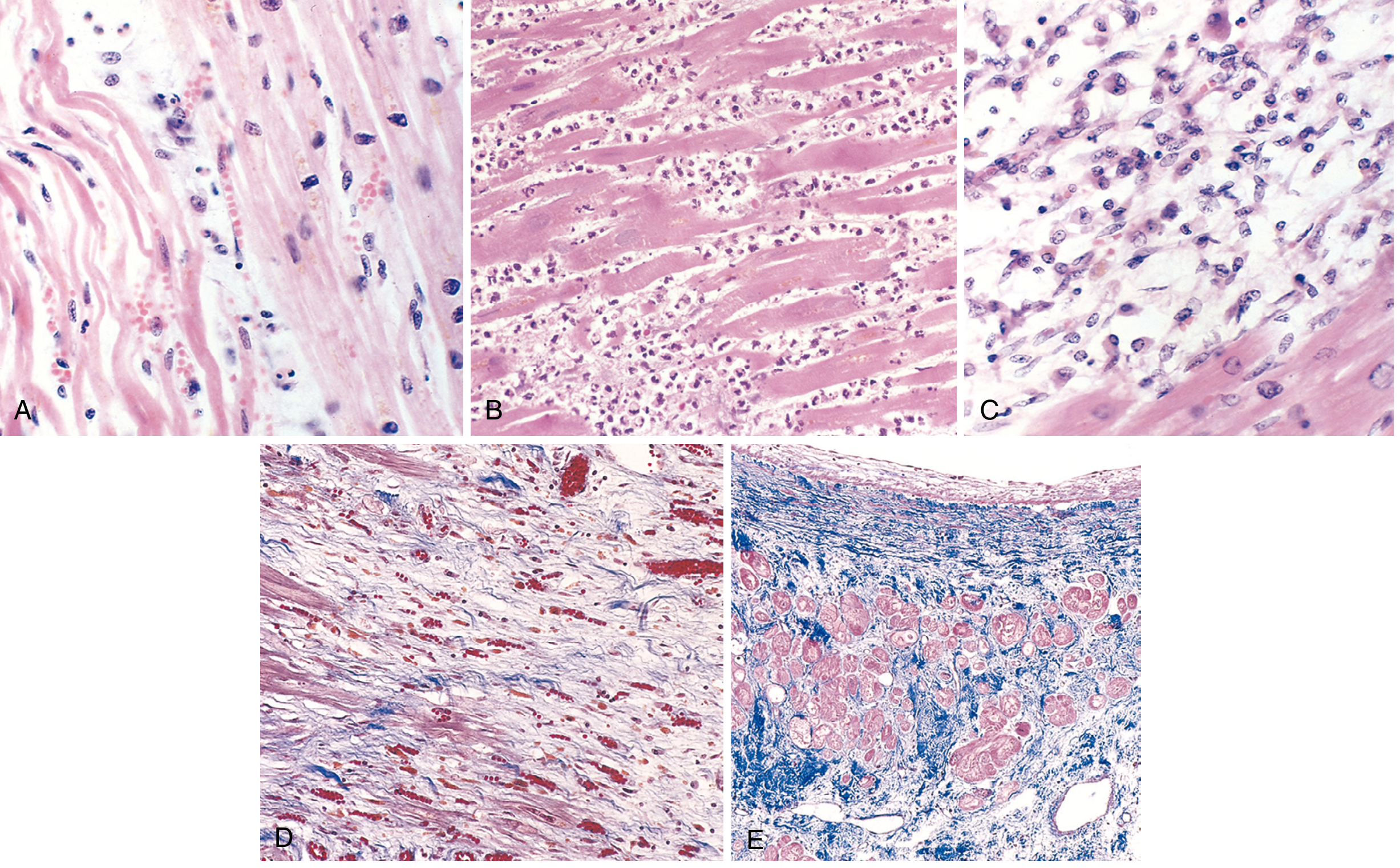
Fig. 12.13 - Microscopic features of MI at progressive time points. A=1 day (wavy fibers), B=3-4 days (neutrophils), C=7-10 days (macrophage phagocytosis), D=granulation tissue, E=healed fibrous scar (Masson trichrome, collagen stains blue). — Robbins, Cotran & Kumar Pathologic Basis of Disease
Phase 1: Early Ischemia (0 to 30 min) - Reversible
- Gross: No changes visible
- Micro: No changes on LM; EM shows myofibril relaxation, glycogen loss, mitochondrial swelling
Phase 2: Irreversible Injury Onset (30 min to 4 hr)
- Gross: Still none (infarcts <12 hrs are invisible grossly without special staining)
- Special stain: Triphenyl tetrazolium chloride (TTC) - viable myocardium stains brick-red (LDH preserved); infarcted area remains pale/unstained (LDH leaked out through damaged membranes)
- Micro: Wavy fibers - viable myocardium contractions stretch and buckle adjacent dead fibers, making them elongated and wavy
- EM: Sarcolemmal disruption; mitochondria develop amorphous densities
Phase 3: Acute Infarct (4-24 hr)
- Gross: Dark mottling (4-12 hr); red-blue discoloration from congestion/extravasation (12-24 hr)
- Micro: Coagulative necrosis - ghost outlines of dead myocytes with:
- Hypereosinophilia of cytoplasm (proteins denatured)
- Pyknosis of nuclei (nuclear condensation)
- Contraction band necrosis at the margins (hypercontracted myofibrils)
- Early neutrophilic infiltrate by 12-24 hr
Phase 4: Neutrophilic/Inflammatory Phase (1-3 days)
- Gross: Yellow-tan center; better demarcation; surrounding hyperemia
- Micro: Complete coagulative necrosis with loss of nuclear detail and striations; brisk, dense neutrophilic infiltrate (most prominent at 1-3 days)
- Risk of cardiac rupture peaks at this phase (softening maximal ~day 10)
Phase 5: Macrophage Phase / Early Granulation (3-10 days)
- Gross: Maximally yellow-tan and soft by days 7-10; depressed red-tan margins
- Micro:
- Neutrophils dying and being replaced by macrophages
- Macrophages phagocytose dead myocytes (most active 3-10 days)
- Granulation tissue (loose collagen + abundant new capillaries) appears at margins by day 7-10
- "Tigroid appearance" may be seen grossly due to red streaks from dilated vascular channels amid yellow necrotic tissue
Phase 6: Granulation Tissue / Collagen Deposition (10-14 days to 2-8 weeks)
- Gross: Red-gray depressed infarct borders (10-14 days); gray-white scar progressively replacing yellow tissue from periphery inward (2-8 weeks)
- Micro: Established granulation tissue with new blood vessels; progressive collagen deposition; cellularity decreases; fibroblasts actively depositing collagen
Phase 7: Healed Fibrous Scar (>6 weeks to 2 months)
- Gross: Gray-white, hard, shrunken fibrous scar ← This is what was found at autopsy in this case
- Well-developed by ~3 months
- Micro: Dense collagenous scar (deep blue on Masson trichrome); residual viable myocytes at margins show compensatory hypertrophy; no inflammatory cells
Summary of Key Gross Features for Age-Dating
| Gross Appearance | Age of Infarct |
|---|---|
| No gross changes | <12-18 hours |
| Dark mottling / red-blue discoloration | 12-24 hours |
| Yellow-tan center, hyperemic border | 1-7 days |
| Maximally yellow, soft, depressed margins | 7-10 days |
| Red-gray depressed borders | 10-14 days |
| Gray-white scar (partial) | 2-8 weeks |
| Complete gray-white fibrous scar | >6 weeks - 3 months ← THIS CASE |
Cause of Sudden Death in This Case
The mechanism of sudden death with an old fibrous scar is:
- Re-entrant ventricular arrhythmia (VT/VF) - scar tissue provides the substrate for electrical re-entry circuits
- New acute MI superimposed on old disease (plaque rupture in a different or same vessel)
- Papillary muscle infarction with acute mitral regurgitation
The old gray-white scar indicates prior ischemic heart disease with healed MI. In ~80% of sudden cardiac deaths from coronary artery disease, there is severe (>75%) stenosis of at least one major coronary artery, with or without a fresh thrombus. Late gadolinium enhancement on MRI (in surviving patients) or fibrosis on autopsy identifies this re-entrant substrate.
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Table 12.5 and pp. 513-515
- The Essentials of Forensic Medicine & Toxicology, 36th ed., pp. 149-150
This is a shared conversation. Sign in to Orris to start your own chat.