Describe and explain Cell death - types, mechanisms, necrosis, apoptosis (basic as contrasted with necrosis), autolysis Ye competency mujhe full detail me samjhao Robbins aur harshmohan ke according Saari sub headings ke saath
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I have comprehensive content from both Robbins books. Now I'll compose the full detailed answer.
Cell Death - Types, Mechanisms, Necrosis, Apoptosis, and Autolysis
(Per Robbins Basic Pathology & Robbins Cotran Pathologic Basis of Disease)
1. OVERVIEW: TYPES OF CELL DEATH
According to Robbins, there are two principal types of cell death: Necrosis and Apoptosis. They differ in their mechanisms, morphology, and roles in physiology and disease.
"Severe mitochondrial damage with depletion of ATP and rupture of lysosomal and plasma membranes are typically associated with necrosis... Apoptosis has many unique features."
- Robbins Cotran Pathologic Basis of Disease
Additionally, autolysis - self-digestion - occurs after cell death and is part of what gives necrosis its ultimate morphologic appearance.
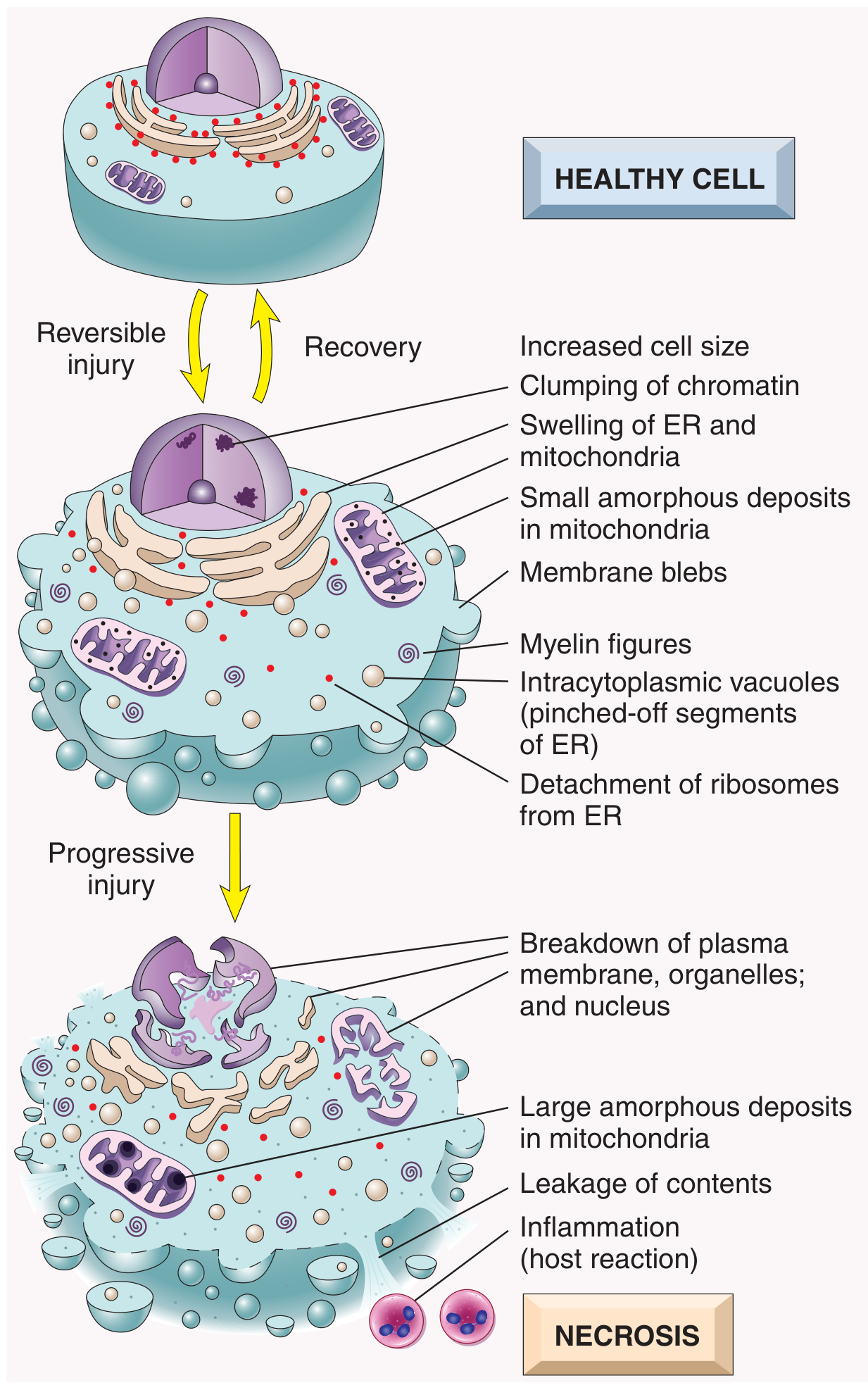
2. SEQUENCE: REVERSIBLE INJURY → IRREVERSIBLE INJURY → CELL DEATH
Before death, a cell passes through a stage of reversible injury, which can recover if the stimulus is removed. The progression to irreversible injury and death is marked by:
- Inability to restore mitochondrial function (oxidative phosphorylation and ATP generation), even after resolution of the original injury
- Loss of membrane integrity - altered structure and loss of function of the plasma membrane and intracellular membranes
- Loss of structural integrity of DNA and chromatin
Key concept (Robbins): "Cellular function may be lost long before cell death occurs, and morphologic changes of cell injury lag behind loss of function and viability." For example, myocardial cells become noncontractile after 1-2 minutes of ischemia but may not die until 20-30 minutes have passed. Morphologic features indicative of myocyte death appear by electron microscopy within 2-3 hours, but are not evident by light microscopy until 6-12 hours later.
3. NECROSIS
3.1 Definition
Necrosis is the form of cell death in which cellular membranes fall apart, cellular enzymes leak out and digest the cell, and there is an accompanying inflammatory reaction. It is typically the result of irreversible cell injury from extrinsic agents (ischemia, toxins, infections, trauma).
Historically described as "accidental" cell death - severe injury irreparably damages so many cellular components that the cell simply "falls apart." However, some forms of necrosis are now known to be genetically regulated (e.g., necroptosis).
3.2 Morphology of Necrosis
Cytoplasmic Changes:
- Increased eosinophilia - due to increased binding of eosin to denatured cytoplasmic proteins and loss of basophilic RNA
- Glassy, homogeneous appearance - due to loss of glycogen particles
- Vacuolated, "moth-eaten" cytoplasm - when enzymes have digested cytoplasmic organelles
- By electron microscopy: discontinuities in plasma and organelle membranes, marked mitochondrial dilation with large amorphous densities, disruption of lysosomes, myelin figures
Nuclear Changes (three patterns, all due to breakdown of DNA and chromatin):
| Nuclear Change | Description |
|---|---|
| Pyknosis | Nuclear shrinkage + increased basophilia; DNA condenses into a dark, shrunken mass |
| Karyorrhexis | The pyknotic nucleus undergoes fragmentation |
| Karyolysis | Basophilia fades due to digestion of DNA by DNase; nucleus dissolves in 1-2 days |
Fate of Necrotic Cells:
- May persist for some time or be digested by enzymes and disappear
- Dead cells may be replaced by myelin figures (phagocytosed or degraded into fatty acids)
- Fatty acids bind calcium - can lead to dystrophic calcification
3.3 Mechanisms Leading to Necrosis
The biochemical basis varies with different injurious stimuli, but common pathways include:
A. Mitochondrial Dysfunction and ATP Depletion
- Failure of oxidative phosphorylation → decreased ATP
- Reduced activity of plasma membrane ATP-dependent sodium pumps → intracellular Na+ accumulation, osmotic water gain → cell swelling and ER dilation
- Compensatory increase in anaerobic glycolysis → lactic acid accumulation → decreased intracellular pH → decreased enzyme activity
- Prolonged depletion: ribosomes detach from rough ER, polysomes dissociate, protein synthesis falls
- Ultimately: irreversible damage to mitochondrial and lysosomal membranes → necrosis
B. Membrane Damage
- Reactive oxygen species (ROS) cause lipid peroxidation
- Decreased phospholipid synthesis (from ATP depletion)
- Increased phospholipid breakdown via Ca2+-activated phospholipases
- Cytoskeletal abnormalities due to Ca2+-activated proteases → membrane detachment and rupture
C. Influx of Intracellular Calcium
- Ischemia and certain toxins → release of Ca2+ from ER and entry from extracellular space
- Activated enzymes: phospholipases (damage membranes), proteases (damage proteins/cytoskeleton), endonucleases (damage DNA), ATPases (hasten ATP depletion)
D. Oxidative Stress (ROS)
- Free radicals with unpaired electrons attack and modify proteins, lipids, carbohydrates, nucleic acids
- Important in ischemia-reperfusion injury, chemical/radiation injury, microbial killing
E. DNA Damage
- Radiation, chemotherapeutic drugs, ROS → DNA damage → p53 activation → either DNA repair or, if irreparable, apoptosis (or necrosis if massive)
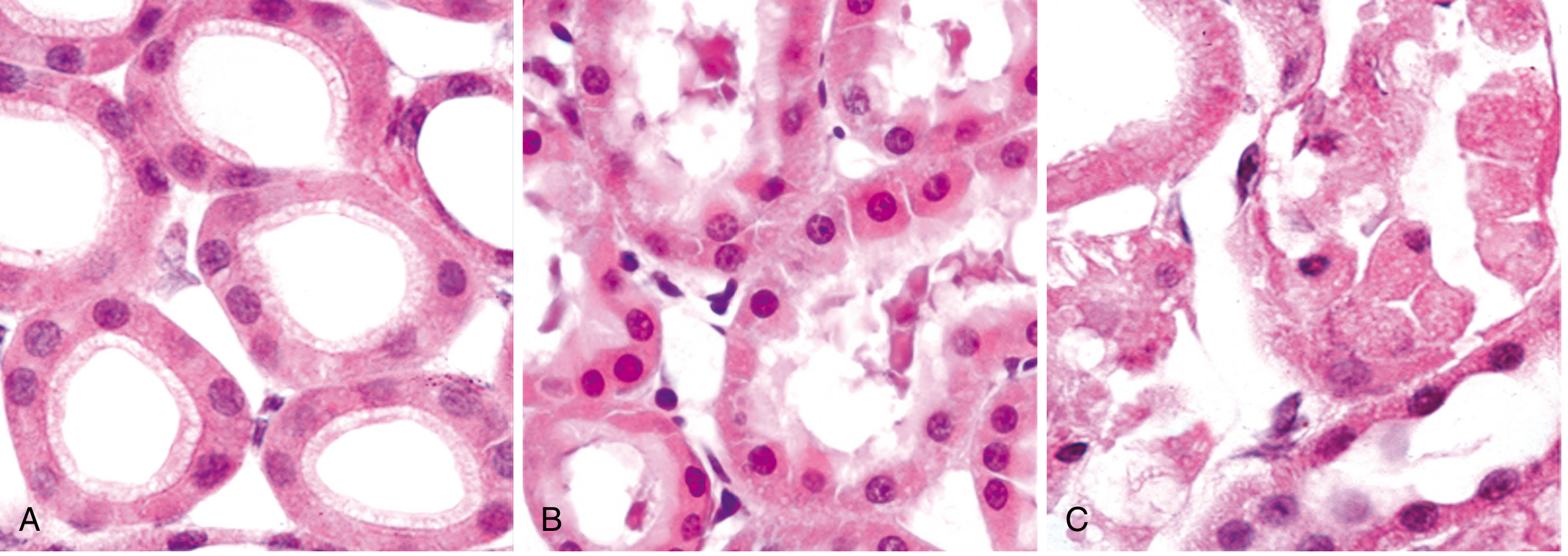
3.4 Morphologic Patterns of Tissue Necrosis
There are 6 morphologic patterns of necrosis recognized by Robbins:
| Pattern | Key Features | Classic Example |
|---|---|---|
| Coagulative necrosis | Architecture preserved; cells are "ghost cells" - outlines remain but nuclei gone. Due to protein denaturation. | Infarcts of heart, kidney, spleen |
| Liquefactive necrosis | Tissue is digested into a liquid, viscous mass. Seen when enzymatic digestion dominates. | Brain infarcts; bacterial abscesses |
| Caseous necrosis | "Cheese-like" (caseum); friable, white-yellow. Necrotic cells form a granular debris enclosed by granuloma. Tissue architecture completely obliterated. | Tuberculosis |
| Fat necrosis | Focal fat destruction; activated lipases split fat cells → free fatty acids + calcium → "chalky white deposits" (saponification) | Acute pancreatitis; traumatic fat necrosis |
| Fibrinoid necrosis | In blood vessel walls; immune complexes + plasma proteins leak into wall → bright pink amorphous material (fibrinoid) | Malignant hypertension; vasculitis; autoimmune diseases |
| Gangrenous necrosis | Not a distinct pattern; usually refers to coagulative necrosis of a limb, often complicated by bacterial infection (wet gangrene) | Ischemic limb; diabetic foot |
4. APOPTOSIS
4.1 Definition
Apoptosis (from Greek: "falling off of leaves") is a form of programmed cell death characterized by activation of defined intracellular molecular pathways. It kills cells with surgical precision, without inflammation or collateral tissue damage.
Robbins describes it as "regulated" cell death - mediated by specific molecular pathways activated under defined circumstances.
4.2 Causes / Situations Where Apoptosis Occurs
Physiologic (normal) situations:
- Programmed cell destruction during embryogenesis (tissue remodeling)
- Hormone-dependent involution (endometrium during menstrual cycle, breast after weaning)
- Cell deletion in proliferating cell populations (intestinal crypt epithelium)
- Elimination of potentially harmful self-reactive lymphocytes
- Cell death after the useful life of the cell (neutrophils after acute inflammation)
Pathologic situations:
- DNA damage - activation of p53 triggers apoptosis when DNA repair fails
- Accumulation of misfolded proteins (ER stress response)
- Certain viral infections (adenovirus, HIV)
- Cytotoxic T-lymphocyte-mediated killing of virally infected and tumor cells
4.3 Morphology of Apoptosis
| Feature | Description |
|---|---|
| Cell size | Reduced (shrinkage) - cells shrink in size, cytoplasm is dense |
| Nucleus | Chromatin condensation and fragmentation into nucleosome-size fragments |
| Plasma membrane | Intact, but altered structure - especially orientation of lipids (phosphatidylserine flipped to outer leaflet) |
| Cellular contents | Intact; released in membrane-bound apoptotic bodies |
| Adjacent inflammation | None - apoptotic bodies are quickly phagocytosed by neighboring cells/macrophages |
| Physiologic role | Often physiologic; eliminates unwanted cells without tissue damage |
4.4 Mechanisms of Apoptosis
Apoptosis results from the activation of enzymes called caspases - proteases containing a cysteine in their active site that cleave proteins after aspartic residues. They exist as inactive proenzymes (procaspases) activated by enzymatic cleavage.
Two phases:
- Initiation phase - some caspases become catalytically active and unleash a cascade
- Execution phase - terminal caspases trigger cellular fragmentation and demise
Two major pathways:
A. Mitochondrial (Intrinsic) Pathway
- Results from increased permeability of the mitochondrial outer membrane → release of pro-apoptotic molecules (cytochrome c) from the intermembrane space into the cytoplasm
- Controlled by the BCL2 family of proteins (named after BCL2, overexpressed in B cell lymphomas):
- Anti-apoptotic: BCL2, BCL-XL, MCL1 (4 BH domains; keep outer mitochondrial membrane impermeable → prevent cytochrome c release)
- Pro-apoptotic: BAX, BAK (BH1-3 domains; oligomerize within outer mitochondrial membrane → enhance permeability)
- BH3-only proteins (sensors of cell stress): sense cellular injury (DNA damage, hypoxia, misfolded proteins) → activate BAX/BAK or inhibit BCL2/BCL-XL → tip balance toward apoptosis
- Cytochrome c released into cytoplasm → binds APAF-1 → forms apoptosome → activates caspase-9 → activates executioner caspases (caspase-3, -7)
B. Death Receptor (Extrinsic) Pathway
- Triggered by "death receptors" on cell surface (members of TNF receptor family: FAS/CD95, TNFR1)
- Ligand binding (FasL/CD95L) → receptor trimerization → recruitment of adaptor proteins (FADD) → binding and activation of procaspase-8 → caspase-8 activation → activates executioner caspases
- Key in: T-cell killing of virally infected cells, elimination of self-reactive lymphocytes
Execution Phase (Common to Both Pathways):
- Activated caspases cleave nuclear lamins (collapse of nuclear structure), cytoskeletal proteins
- Endonucleases fragment DNA into oligonucleosomal fragments (ladder pattern on gel)
- Cells break up into apoptotic bodies - membrane-enclosed fragments containing cytoplasm and organelles
- Apoptotic bodies display "eat-me" signals (phosphatidylserine on outer leaflet, thrombospondin) → rapidly phagocytosed by macrophages and neighboring cells → no inflammatory response
5. APOPTOSIS vs NECROSIS - COMPARISON TABLE
| Feature | Necrosis | Apoptosis |
|---|---|---|
| Cell size | Enlarged (swelling) | Reduced (shrinkage) |
| Nucleus | Pyknosis → karyorrhexis → karyolysis | Fragmentation into nucleosome-size fragments |
| Plasma membrane | Disrupted | Intact; altered lipid orientation |
| Cellular contents | Enzymatic digestion; leak out of cell | Intact; released in apoptotic bodies |
| Adjacent inflammation | Frequent (always present) | No |
| Process type | Passive, "accidental" (mostly) | Active, programmed (energy-requiring) |
| Energy requirement | No (passive process) | Yes (requires ATP) |
| Caspase activation | No | Yes |
| DNA fragmentation | Random, non-specific | Internucleosomal (specific, ladder pattern) |
| Physiologic/pathologic role | Usually pathologic (culmination of irreversible injury) | Often physiologic; also pathologic (DNA damage, viral infections) |
(Robbins Cotran, Table 2.2)
6. AUTOLYSIS
6.1 Definition
Autolysis (Greek: autos = self, lysis = dissolution) is self-digestion of the cell by its own enzymes, particularly those released from ruptured lysosomes. It is a post-mortem (or post-necrotic) process and is a component of what creates the final morphologic picture of necrosis.
6.2 Mechanism
When a cell is irreversibly injured and dies (especially by necrosis):
- Lysosomal membranes rupture → release of hydrolytic enzymes (proteases, lipases, DNases, RNases, glycosidases) into the cytoplasm
- These enzymes are active at low pH - the acidic intracellular environment favors their activity
- The enzymes digest intracellular organelles, proteins, lipids, and DNA
- This is compounded by enzymes released from invading leukocytes recruited by the inflammatory response
6.3 Autolysis vs Heterolysis
- Autolysis = digestion by the cell's own enzymes (from its lysosomes)
- Heterolysis = digestion by enzymes derived from leukocytes (neutrophils and macrophages) that arrive in the inflammatory reaction
Both occur together in necrosis to create the ultimate dissolution of the tissue.
6.4 Autolysis in Morphologic Patterns of Necrosis
- Coagulative necrosis: Protein denaturation outpaces autolysis - cell outlines are preserved ("ghost cells")
- Liquefactive necrosis: Autolysis/enzymatic digestion dominates - tissue is completely liquefied (classic in brain infarcts and abscesses)
6.5 Post-Mortem Autolysis (Forensic Context)
After death, in the absence of blood flow and immune defense, autolysis continues unchecked throughout the body - this is the basis of post-mortem tissue changes and is an important concept in forensic pathology and tissue banking.
7. OTHER FORMS OF CELL DEATH (Brief Mention per Robbins Cotran)
While necrosis and apoptosis are the two principal forms, Robbins also identifies:
- Necroptosis ("programmed necrosis") - morphologically resembles necrosis but is mediated by defined molecular signals (RIPK1, RIPK3); triggered by TNF, viral infections; causes inflammation like necrosis
- Pyroptosis - associated with inflammasome activation; mediated by caspase-1/11; releases IL-1β; occurs in inflammatory responses to pathogens
- Ferroptosis - iron-dependent form of regulated cell death mediated by lipid peroxidation; not mediated by caspases
SUMMARY FLOWCHART
CELL INJURY
|
├── MILD / SHORT DURATION → REVERSIBLE INJURY (cellular swelling, fatty change)
| ↕ recovery
└── SEVERE / PROLONGED → IRREVERSIBLE INJURY
|
┌─────────────┴──────────────┐
↓ ↓
NECROSIS APOPTOSIS
(accidental, passive) (programmed, active)
Membrane disruption Membrane intact
Enzyme leakage Apoptotic bodies
INFLAMMATION present No inflammation
Pyknosis/karyorrhexis Chromatin fragmentation
Autolysis/heterolysis Phagocytosed cleanly
Sources:
- Robbins & Kumar Basic Pathology, 11th Ed., Ch. 1 - Cell Injury, Cell Death, and Adaptations
- Robbins, Cotran & Kumar Pathologic Basis of Disease, 10th Ed., Ch. 2 - Cellular Responses to Stress and Toxic Insults (Table 2.2, Figs. 2.8-2.16)
Note (Harshmohan): The library currently contains only the Robbins titles. The core content on necrosis, apoptosis, and autolysis is consistent across Harshmohan's Textbook of Pathology - the same morphologic patterns (coagulative, liquefactive, caseous, fat, fibrinoid, gangrenous necrosis) and apoptosis pathways are described using the same classification. Harshmohan additionally emphasizes the role of lysosomes in autolysis, the concept of "somatic death" vs "cell death", and gives extra weight to the practical differential of autolysis (post-mortem change) from ante-mortem necrosis in histopathology.
This is a shared conversation. Sign in to Orris to start your own chat.