Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome" AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathophysiology mechanism alveolar damage
I have rich content from the textbooks and excellent images. I can now write a comprehensive, well-structured answer on the mechanism of ARDS.
Acute Respiratory Distress Syndrome (ARDS) — Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by severe lung inflammation, characterised by bilateral infiltrates, hypoxemia (PaO₂/FiO₂ < 300 on ≥5 cmH₂O PEEP), and decreased lung compliance, developing within 7 days of a known insult. The Berlin definition (2012) stratifies severity: mild (P/F 200–300), moderate (100–200), and severe (<100).
1. Initiating Insults
ARDS arises from either direct (pulmonary) or indirect (extrapulmonary) lung injury:
| Direct (pulmonary) | Indirect (systemic) |
|---|---|
| Pneumonia (bacterial, viral) | Sepsis (most common overall cause) |
| Aspiration of gastric contents | Severe trauma / shock |
| Toxic inhalation / near drowning | Pancreatitis |
| Pulmonary contusion | Massive transfusion (TRALI) |
2. The Alveolar-Capillary Membrane Breakdown
The central event in ARDS is disruption of the alveolar-capillary barrier, which consists of:
- The capillary endothelium
- The basement membrane
- The alveolar epithelium (type I and type II pneumocytes)
Under normal conditions this barrier prevents movement of proteins and fluid into the alveolar space. In ARDS, injury to either surface — or both — leads to a high-permeability pulmonary edema, fundamentally distinct from the hydrostatic (cardiogenic) edema of heart failure.
The epithelial surface is particularly important. Type I alveolar epithelial cells cover ~95% of the alveolar surface and are highly vulnerable to injury. Type II pneumocytes, which produce surfactant and can regenerate type I cells, are more resistant but are also impaired.
3. Neutrophil Recruitment and Activation — The Core Driver
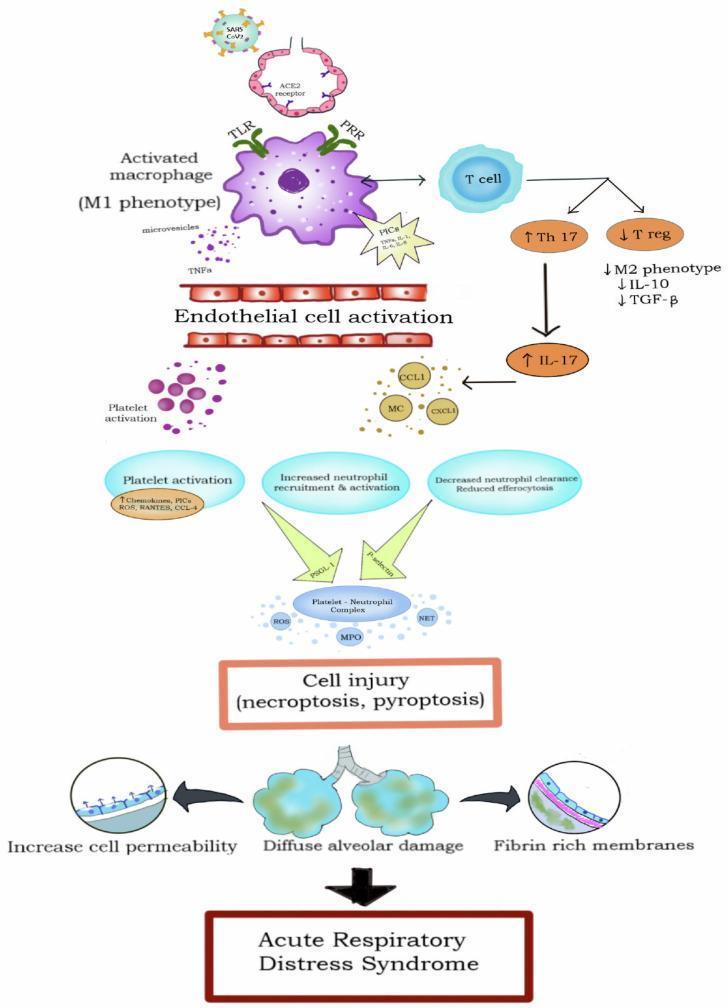
Neutrophils are the primary effector cells of ARDS lung injury. The sequence:
- Systemic activation: The initiating insult activates the innate immune system — pattern-recognition receptors (TLRs, PRRs) on macrophages detect DAMPs (damage-associated molecular patterns) or PAMPs (pathogen-associated patterns).
- Cytokine storm: Activated alveolar macrophages release pro-inflammatory cytokines — TNF-α, IL-1β, IL-6, IL-8 — into the alveolar space and systemic circulation.
- Neutrophil margination and sequestration: IL-8 and other chemokines recruit neutrophils into the pulmonary capillaries. Neutrophils become stiff, sequester in capillary beds, and adhere to activated endothelium via upregulated adhesion molecules (ICAM-1, selectins).
- Transendothelial and transepithelial migration: Neutrophils migrate into the interstitium and alveolar space.
- Toxic mediator release: Activated neutrophils release:
- Reactive oxygen species (ROS) — oxidative damage to lipid membranes
- Proteases (elastase, matrix metalloproteinases, cathepsins) — degrade the extracellular matrix and tight junctions
- Neutrophil extracellular traps (NETs) — webs of chromatin and enzymes that amplify injury
- Myeloperoxidase (MPO) — generates hypochlorous acid
- Barrier destruction: Together these mediators destroy tight junction proteins (occludin, claudins, ZO-1) of both endothelium and epithelium, producing the high-permeability leak.
Key finding: BAL fluid from ARDS patients contains a high neutrophil count (often >80% PMNs), whereas normal BAL has <5%.
4. Surfactant Dysfunction
Type II pneumocytes are injured, reducing surfactant synthesis. Surfactant already present is:
- Diluted by protein-rich edema fluid
- Inactivated by phospholipase A2 (released from activated neutrophils and pancreatic/systemic sources) — phospholipase A2 enzymatically degrades surfactant phospholipids
- Oxidised by ROS
The result is markedly elevated alveolar surface tension, leading to widespread alveolar collapse (atelectasis) and further reduction in lung compliance. This explains the characteristic "baby lung" phenomenon — only a small fraction of alveoli remain recruitable.
5. Phases of Histopathological Injury: Diffuse Alveolar Damage (DAD)
The pathological correlate of ARDS is Diffuse Alveolar Damage (DAD), which evolves in phases:
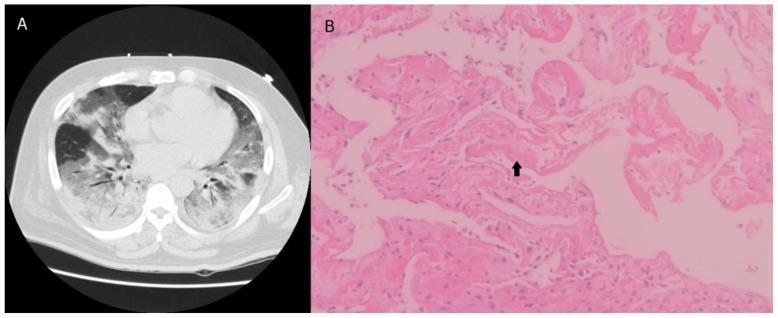
Exudative phase (0–7 days)
- Protein-rich, fibrin-containing alveolar edema
- Sloughing of type I pneumocytes — the alveolar basement membrane is denuded
- Formation of hyaline membranes — eosinophilic deposits of fibrin, necrotic cell debris, and protein lining the alveolar walls (pathognomonic of DAD)
- Neutrophilic infiltration of the interstitium and alveolar space
- Capillary microthrombi formation
Proliferative phase (7–21 days)
- Type II pneumocyte hyperplasia — attempt to repopulate denuded epithelium
- Fibroblast proliferation and early fibrosis begins
- Macrophage-mediated clearance of debris
- Restoration of surfactant production if recovery proceeds
Fibrotic phase (>21 days, in some patients)
- Permanent fibrotic remodelling of alveoli
- Loss of lung architecture, reduced compliance
- Not all patients progress to this phase — good recovery is possible
6. Coagulation and Microvascular Thrombosis
ARDS generates a procoagulant state in the alveolar microenvironment:
- Damaged endothelium exposes tissue factor → activates the extrinsic coagulation cascade
- Fibrin deposited within capillaries and alveolar spaces
- Plasminogen activator inhibitor-1 (PAI-1) is elevated → suppresses fibrinolysis
- Capillary microthrombi worsen ventilation-perfusion (V/Q) mismatch and contribute to pulmonary hypertension
7. Impaired Alveolar Fluid Clearance
Under normal conditions, Na⁺-K⁺-ATPase pumps on type II and type I pneumocytes actively reabsorb sodium (and osmotically water) from the alveolar space. In ARDS:
- Injury to type I and II cells impairs active Na⁺ transport
- Protein-rich edema accumulates and cannot be cleared
- Catecholamines, which normally upregulate Na⁺ transport, lose their effect in the injured epithelium
The rate of alveolar fluid clearance is an independent predictor of outcome: faster clearance correlates with lower mortality.
8. Angiopoietins and Vascular Dysregulation
The angiopoietin-Tie2 axis regulates endothelial barrier function:
- Angiopoietin-1 (Ang-1): stabilises endothelial junctions — protective
- Angiopoietin-2 (Ang-2): released from Weibel-Palade bodies during endothelial activation → destabilises junctions, promotes vascular leak and upregulates inflammatory adhesion molecules
Elevated plasma Ang-2 levels are associated with increased ARDS severity and mortality.
9. Physiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + collapse | Intrapulmonary shunt → refractory hypoxemia |
| Surfactant loss + edema | Reduced compliance (stiff lungs) |
| Capillary microthrombi + hypoxic vasoconstriction | V/Q mismatch, pulmonary hypertension |
| Inflammatory cytokine spill-over | Multi-organ dysfunction |
| Mechanical ventilation (high volumes/pressures) | Ventilator-induced lung injury (VILI) — biotrauma amplifies inflammation |
10. Two Biological Phenotypes
Large ARDS trial data (latent class analysis) has identified at least two phenotypes:
- Hyperinflammatory (phenotype 2): high IL-6, IL-8, Ang-2, low protein C; higher mortality (~40%), may respond to higher PEEP and statin therapy
- Hypoinflammatory (phenotype 1): milder biomarker elevation, lower mortality (~20%)
This heterogeneity partly explains why no single pharmacological therapy has shown consistent mortality benefit.
Summary Pathway
Systemic insult (sepsis, trauma, aspiration…)
↓
Innate immune activation → macrophage cytokine release (TNF-α, IL-1β, IL-8)
↓
Neutrophil sequestration in pulmonary capillaries
↓
ROS, proteases, NETs → Alveolar-capillary barrier destruction
↓
High-permeability edema floods alveoli
↓
Surfactant inactivation → alveolar collapse
Impaired Na⁺ transport → edema accumulation
Procoagulant state → microvascular thrombosis
↓
DAD: hyaline membranes, type I cell sloughing, type II hyperplasia
↓
Severe hypoxemia (refractory to O₂), decreased compliance, pulmonary HTN
↓
Respiratory failure → mechanical ventilation required
Sources:
- Murray & Nadel's Textbook of Respiratory Medicine — Chapter 134 (Etiology and Pathogenesis of ARDS)
- Murray & Nadel's Textbook of Respiratory Medicine — Chapter 94 (Diffuse Alveolar Damage)
- Goldman-Cecil Medicine — Chapter 91 (ARDS)
- Sabiston Textbook of Surgery — Critical Care chapter
This is a shared conversation. Sign in to Orris to start your own chat.