You are 1st year resident of physiology presenting a seminar on topic " Pathophysiology of fever". Give me answer for the following questions: How to differentiate between hyperthermia and fever Types n egs of pyrogens Why are chills seen in fever What is defervescence and its moa How nsaids reduce fever Metabolic effects of fever How is fever beneficial and harmful Febrile seizures
Seminar: Pathophysiology of Fever
1. Fever vs. Hyperthermia - How to Differentiate
| Feature | Fever | Hyperthermia |
|---|---|---|
| Definition | Regulated elevation of the hypothalamic set point | Unregulated rise in body temp due to failure to dissipate heat |
| Mechanism | Pyrogen → PGE2 → ↑ set point → body works to reach the new set point | Heat gain exceeds body's cooling capacity; set point is NORMAL |
| Hypothalamic set point | Raised | Normal / unchanged |
| Response to antipyretics (NSAIDs) | Effective - reduces fever | Ineffective - does not help |
| Vasoconstriction/shivering | Present initially (to raise temperature to new set point) | Absent - body is actively trying to lose heat (vasodilation, sweating) |
| Core temp > 41°C | Possible in severe sepsis | Common - most cases of temp >41°C (105.8°F) are hyperthermia |
| Examples | Bacterial/viral infection, malignancy, autoimmune | Heat stroke, malignant hyperthermia, NMS, thyroid storm |
Key teaching point: In hyperthermia, the thermoregulatory system is overwhelmed or bypassed - the hypothalamus WANTS to cool down but cannot. In fever, the hypothalamus deliberately raises the set point.
- Rosen's Emergency Medicine, Concepts and Clinical Practice, p. 123
2. Pyrogens - Types and Examples
A. Exogenous Pyrogens
| Type | Examples |
|---|---|
| Bacterial products | Lipopolysaccharide (LPS/endotoxin) from gram-negative bacteria - the classic exogenous pyrogen |
| Gram-positive products | Peptidoglycan, lipoteichoic acid, superantigens (TSST-1, Staphylococcal enterotoxins) |
| Viral products | Double-stranded RNA, viral capsid proteins |
| Fungal | Zymosan, mannans |
| Drug-related | Blood products, vaccines, certain medications |
- Gene expression and synthesis of pyrogenic cytokines (endogenous pyrogens)
- Direct induction of cyclooxygenase-2 (COX-2) via TLR-4 signaling → PGE2 production
B. Endogenous Pyrogens
| Cytokine | Source |
|---|---|
| IL-1β (Interleukin-1 beta) | Macrophages, monocytes - the classic endogenous pyrogen |
| TNF-α (Tumor Necrosis Factor-alpha) | Macrophages, T cells |
| IL-6 (Interleukin-6) | Macrophages, endothelium |
| IFN-γ (Interferon-gamma) | T lymphocytes, NK cells |
"Substances that induce fever are called pyrogens and include bacterial products (exogenous pyrogens, e.g., LPS) and cytokines, principally IL-1 and TNF (called endogenous pyrogens)."
- Robbins, Cotran & Kumar - Pathologic Basis of Disease
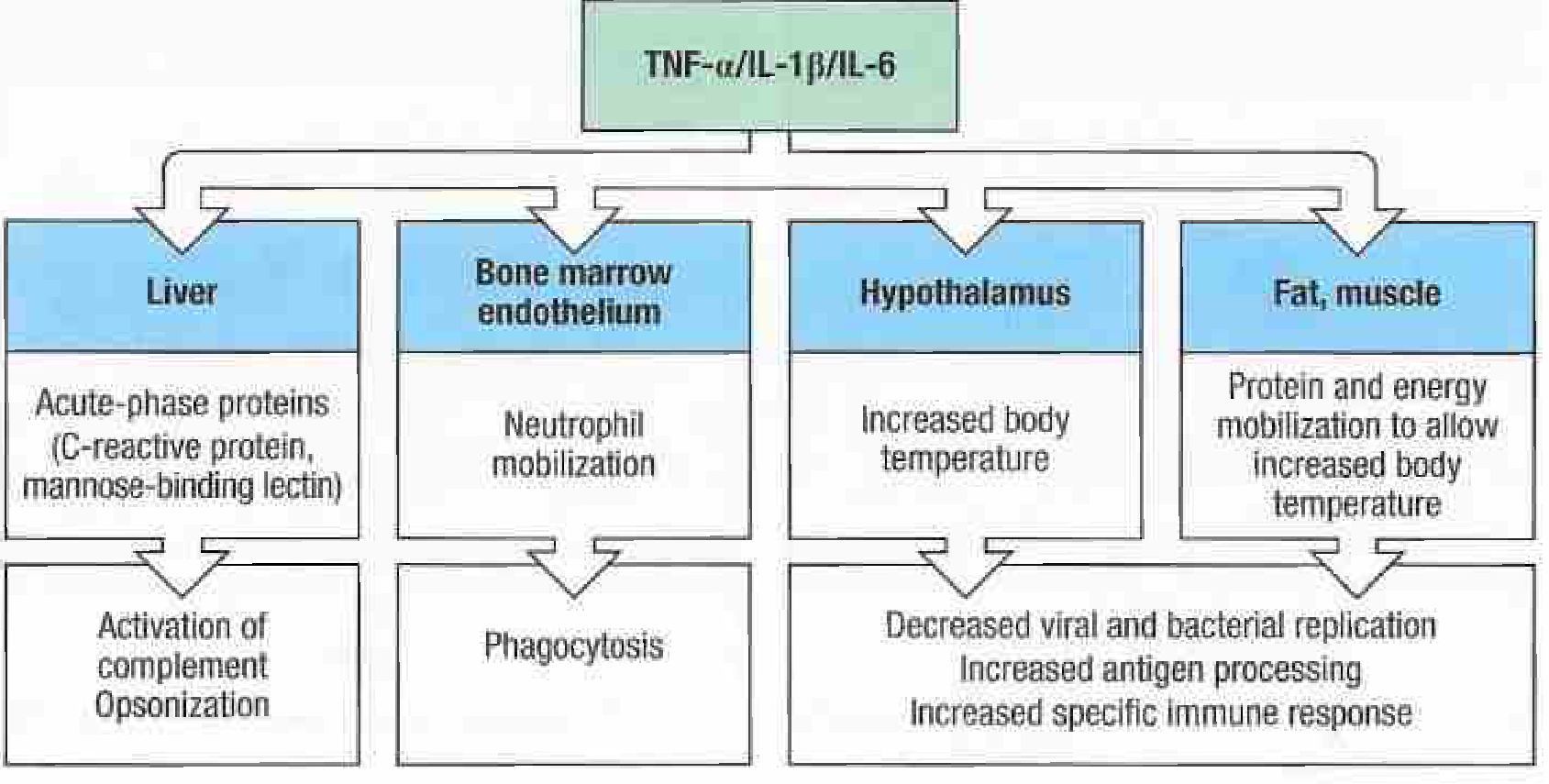
3. Why Are Chills Seen in Fever?
-
Pyrogenic cytokines (IL-1, TNF-α, IL-6) reach the hypothalamus.
-
PGE2 is synthesized → the hypothalamic thermostat set point is raised (e.g., from 37°C to 39°C).
-
At this point, the actual body temperature is still 37°C - the body perceives itself as too cold relative to the new set point.
-
The hypothalamus activates heat-conserving and heat-generating mechanisms to bring the actual temperature up to the new set point:
- Peripheral vasoconstriction → reduced heat loss through the skin → patient feels cold, skin becomes pale/mottled
- Piloerection ("goosebumps") → conserves heat (vestigial mechanism)
- Shivering (rigors) → involuntary rhythmic skeletal muscle contractions → generates heat through increased metabolic rate
- Behavioral changes → patient seeks warmth, curls up, wraps in blankets
- ↑ Brown fat catabolism → non-shivering thermogenesis
-
Once the actual body temperature reaches the new set point, chills stop and the patient feels hot.
"PGE2 raises the set point of the temperature range by a combination of effects, including peripheral vasoconstriction, increased metabolic heat production, shivering, and behavioral changes that conserve heat."
- Rosen's Emergency Medicine, p. 123
4. Defervescence and Its Mechanism
Two Patterns of Defervescence:
| Pattern | Description | Clinical significance |
|---|---|---|
| Crisis (Lysis by crisis) | Sudden drop in temperature, accompanied by profuse sweating and vasodilation | Seen in lobar pneumonia (historically), malaria |
| Lysis (Gradual lysis) | Slow, gradual fall in temperature over hours to days | More common; seen in most viral and bacterial infections |
Mechanism of Action of Defervescence:
- Pyrogenic cytokines decrease → PGE2 levels fall in the hypothalamus.
- The set point returns to normal (37°C).
- Now the actual body temperature (still elevated at, say, 39°C) is above the new normal set point.
- The hypothalamus activates heat-dissipating mechanisms:
- Cutaneous vasodilation → increased heat loss through the skin → patient appears flushed/red
- Profuse sweating → evaporative heat loss
- Behavioral changes → removes blankets, seeks cool environment
- Body temperature gradually falls back to normal.
5. How NSAIDs Reduce Fever
Step-by-step mechanism:
- Exogenous pyrogens (e.g., LPS) or inflammatory stimuli activate immune cells → release of IL-1β, TNF-α, IL-6
- These endogenous pyrogens act on the hypothalamic vasculature → upregulate Cyclooxygenase-2 (COX-2)
- COX-2 converts arachidonic acid to PGE2 (Prostaglandin E2)
- PGE2 binds to EP1 and EP3 receptors in the preoptic area of the hypothalamus → raises temperature set point
- NSAIDs (e.g., ibuprofen, aspirin, naproxen) → competitively and irreversibly (aspirin) or reversibly (other NSAIDs) inhibit COX-1 and COX-2
- ↓ PGE2 synthesis → set point falls back to normal → defervescence occurs
"NSAIDs, including aspirin, reduce fever by inhibiting prostaglandin synthesis."
- Robbins, Cotran & Kumar - Pathologic Basis of Disease
"Medications such as nonsteroidal anti-inflammatory drugs and aspirin block the activity of enzymes involved in the synthesis of prostaglandin E2."
- Goldman-Cecil Medicine
6. Metabolic Effects of Fever
| Metabolic Effect | Detail |
|---|---|
| ↑ Basal Metabolic Rate | ~10-13% per °C rise; increased O2 consumption and CO2 production |
| ↑ Protein catabolism | Muscle protein is broken down; negative nitrogen balance; explains weight loss and weakness in prolonged fever |
| Gluconeogenesis | Hepatic gluconeogenesis is activated (driven by IL-1 and TNF-α); glucose is needed as fuel |
| Fat mobilization | Lipolysis increases; brown fat catabolism increases (non-shivering thermogenesis) |
| Acute-phase response | IL-6 drives the liver to produce C-reactive protein (CRP), fibrinogen, serum amyloid A, mannose-binding lectin - all part of innate immune defense |
| ↓ Albumin and transferrin | These are "negative acute-phase reactants" - their production is reduced |
| Iron sequestration | Lactoferrin and ferritin increase; serum iron and zinc fall - this is a bacteriostatic mechanism (many bacteria need iron to grow) |
| ↑ Heart rate | ~2-5 beats/min per 1°F rise in temperature |
| ↑ Respiratory rate | Increased O2 demand drives tachypnea |
| Water/electrolyte loss | Sweating during defervescence causes volume depletion |
7. How Fever is Beneficial and Harmful
Beneficial Effects of Fever
| Benefit | Mechanism |
|---|---|
| Inhibits microbial replication | Most pathogens replicate optimally at 37°C; elevated temperature impairs their growth and virulence; shown clearly in amphibians and some mammals |
| Enhanced phagocytosis | Neutrophil and macrophage activity increases at higher temperatures |
| Increased neutrophil mobilization | TNF-α, IL-1 drive accelerated release of granulocytes from bone marrow |
| Enhanced lymphocyte proliferation | T- and B-cell responses are augmented |
| Improved antigen processing | Heat stress enhances antigen presentation by dendritic cells |
| Acute-phase protein production | CRP, MBL, fibrinogen, SAA act as opsonins and activate complement |
| Iron sequestration | Reduces available iron for bacterial growth (bacteriostatic) |
"An elevated body temperature has been shown to help amphibians ward off microbial infections, and it is possible that fever is a protective host response in mammals as well."
- Robbins, Cotran & Kumar
"When patients fail to develop a fever despite severe bacterial infection, morbidity and mortality tend to be higher."
- Goldman-Cecil Medicine
Harmful Effects of Fever
| Harm | Mechanism |
|---|---|
| Febrile seizures | Especially in children 6 months - 5 years; rapid rise in temperature lowers seizure threshold |
| Increased metabolic demand | Dangerous in patients with cardiac failure, respiratory compromise, or anemia |
| Protein catabolism | Prolonged fever leads to muscle wasting and negative nitrogen balance |
| Dehydration | Insensible losses via sweating; dangerous in elderly and infants |
| CNS effects | High fever (>41°C) causes confusion, delirium, coma |
| Cardiovascular stress | Tachycardia increases cardiac workload; dangerous in ischemic heart disease |
| Febrile status epilepticus | Can cause hippocampal injury and temporal lobe epilepsy later in life |
| Hyperthermia (if uncontrolled) | Temperatures >41-42°C can cause direct protein denaturation, cellular injury, multi-organ failure |
8. Febrile Seizures
Definition
Classification
| Type | Features |
|---|---|
| Simple febrile seizure | Generalized (tonic-clonic), <15 minutes, single episode within 24 hours, no focal features, full recovery |
| Complex febrile seizure | ONE or more of: duration >15 min, focal features (focal ictal activity or Todd's palsy), >1 seizure in 24 hours |
Why Does Fever Cause Seizures?
- Rapid rise in temperature (not necessarily very high temperature) is the key trigger
- Fever increases neuronal excitability: depolarizes neuronal membranes, alters ion channel kinetics, increases synaptic transmission
- The developing brain (myelination incomplete) has a lower seizure threshold compared to adults
- Genetic predisposition plays a role (polygenic inheritance; some families show autosomal dominant transmission with mutations in SCN1A sodium channels)
Recurrence Risk
- 30-40% of children will have at least one recurrence
- Predictors of recurrence: age <1 year at first seizure, family history of febrile seizures/epilepsy, daycare attendance (↑ febrile illnesses)
Risk of Later Epilepsy
- Risk of afebrile epilepsy by age 25: ~7% (vs. ~1% in general population)
- Risk increases with complex features:
- 1 complex feature: 6-8%
- 2 complex features: 17-22%
- All 3 complex features: ~49%
- Febrile status epilepticus can injure the hippocampus → hippocampal sclerosis → temporal lobe epilepsy
Genetic Associations
- Dravet syndrome (SMEI) - truncating mutation in SCN1A; presents with febrile seizures in infancy, progresses to afebrile seizures and cognitive decline
- Other genetic epilepsy syndromes can begin with febrile seizures
Management
- Acute: Diazepam (rectally or IV) to terminate prolonged seizures
- Prophylaxis: Chronic anti-seizure drugs are generally NOT recommended (side effects outweigh benefit for simple febrile seizures)
- For high-risk children: Rectal diazepam at time of fever to prevent recurrence (avoids chronic drug exposure)
- Chronic phenobarbital prophylaxis: Shown to have substantial cognitive side effects (Farwell et al., 1990) - not recommended
Summary Table: The Fever Cascade
Infection/Inflammation/Neoplasm
↓
Immune cells (macrophages, monocytes)
↓
Exogenous pyrogens → Endogenous pyrogens (IL-1β, TNF-α, IL-6, IFN-γ)
↓
Preoptic anterior hypothalamus
↓
↑ COX-2 → ↑ PGE2 (EP1/EP3 receptors)
↓
RAISED THERMOSTAT SET POINT
↓
Heat conservation: vasoconstriction + shivering (CHILLS)
↓
Core temp reaches new set point → FEVER maintained
↓
Cause resolves / NSAIDs → ↓ PGE2 → set point normalizes
↓
Heat dissipation: vasodilation + sweating (DEFERVESCENCE)
- Rosen's Emergency Medicine - Concepts and Clinical Practice (Pathophysiology of Fever, p. 123)
- Robbins, Cotran & Kumar - Pathologic Basis of Disease (Systemic Effects of Inflammation)
- Goldman-Cecil Medicine, International Edition (Pathobiology of Fever)
- Janeway's Immunobiology, 10e (Acute-Phase Response)
- Goodman & Gilman's Pharmacological Basis of Therapeutics (Febrile Convulsions)
- Bradley and Daroff's Neurology in Clinical Practice (Febrile Seizures)
suggest some questions the professors can ask the same topic with answers
Viva Questions: Pathophysiology of Fever
BASIC / OPENING QUESTIONS
MECHANISM-BASED QUESTIONS
- Aspirin (acetylsalicylic acid): Causes irreversible acetylation of the serine residue in the active site of both COX-1 and COX-2. New enzyme must be synthesized to restore function. In platelets (which lack nuclei), COX-1 inhibition is permanent for the platelet's lifetime (~10 days).
- Ibuprofen: Causes reversible, competitive inhibition of both COX isoforms. Effect wears off as the drug is cleared.
- Both reduce fever by the same endpoint: ↓ PGE2 → set point normalizes.
TRICKY / APPLIED QUESTIONS
- No ceiling mechanism: In fever, the body's own anti-pyretic mechanisms (endogenous antipyretics like arginine vasopressin, α-MSH, IL-10) and the fixed dose of pyrogen limit how high the temperature rises - usually not beyond 41°C in true fever. In hyperthermia, there is no such regulatory ceiling; temperatures can exceed 42-43°C.
- No adaptive response: Fever is a coordinated response where the body "wants" the higher temperature. In hyperthermia, the hypothalamus is desperately trying to cool down but cannot. Temperatures above 41-42°C cause direct protein denaturation, enzyme dysfunction, and multi-organ failure.
- Arginine vasopressin (AVP) - released from hypothalamus; acts centrally to limit fever
- α-Melanocyte-stimulating hormone (α-MSH) - potent antipyretic and anti-inflammatory peptide
- IL-10 - anti-inflammatory cytokine; inhibits macrophage activation and cytokine production
- Glucocorticoids - cortisol suppresses cytokine production and PGE2 synthesis These constitute a negative feedback system - as fever rises, anti-inflammatory signals also rise to keep the response proportionate.
- The upper limit of fever is controlled by endogenous antipyretics (AVP, α-MSH, IL-10) - these kick in as temperature rises, preventing runaway fever.
- Febrile seizures depend not so much on the absolute temperature but on the rate of temperature rise - a rapid rise from 37°C to 38.5°C can trigger a seizure just as readily as a higher temperature. The immature, incompletely myelinated brain of young children has a lower seizure threshold and is more sensitive to rapid temperature changes and their effects on ion channel kinetics.
- 1 complex feature (prolonged >15 min, focal, or >1 in 24h): 6-8%
- 2 complex features: 17-22%
- All 3 complex features: ~49%
- Pre-existing neurodevelopmental abnormalities
- Family history of epilepsy
- Febrile status epilepticus (can cause hippocampal injury → sclerosis → temporal lobe epilepsy)
- Recurrent febrile seizures
- TNF-α and IL-1 drive accelerated release of granulocytes from the bone marrow (explaining the early neutrophilia with left shift)
- IL-6 is integral to the production of acute-phase reactants (CRP, fibrinogen, haptoglobin, serum amyloid A)
- Prolonged infection → colony-stimulating factors (CSFs) from macrophages → proliferation of precursors in bone marrow
- Typhoid fever (Salmonella typhi)
- Leptospirosis
- Rickettsiosis
- Dengue fever
- Legionellosis
- Babesiosis
CLASSIC TRAP / EXAMINER FAVOURITE
- Overwhelming infection with immune exhaustion
- Elderly or immunocompromised patient (blunted cytokine response)
- Antipyretic medication masking fever
- Associated with development of septic shock and worse outcomes
Create a study guide PDF with all viva Q&A for quick review
ls -la /home/daytona/workspace/ 2>/dev/null || echo "workspace empty or not found"
mkdir -p /home/daytona/workspace/fever-study-guide && python3 -c "from reportlab.lib import colors; print('reportlab ok')"
~/fever-study-guide/generate_pdf.py
from reportlab.lib.pagesizes import A4
from reportlab.lib import colors
from reportlab.lib.styles import getSampleStyleSheet, ParagraphStyle
from reportlab.lib.units import cm
from reportlab.platypus import (
SimpleDocTemplate, Paragraph, Spacer, Table, TableStyle,
HRFlowable, PageBreak, KeepTogether
)
from reportlab.lib.enums import TA_LEFT, TA_CENTER, TA_JUSTIFY
from reportlab.platypus import BalancedColumns
OUTPUT = "/home/daytona/workspace/fever-study-guide/Fever_Viva_Study_Guide.pdf"
# ── Colour palette ──────────────────────────────────────────────────────────
NAVY = colors.HexColor("#1a2e4a")
TEAL = colors.HexColor("#0d7377")
AMBER = colors.HexColor("#e8a020")
LIGHT_BG = colors.HexColor("#f0f6fb")
Q_BG = colors.HexColor("#dff0ea")
WARN_BG = colors.HexColor("#fff3cd")
RED = colors.HexColor("#c0392b")
WHITE = colors.white
# ── Document ─────────────────────────────────────────────────────────────────
doc = SimpleDocTemplate(
OUTPUT,
pagesize=A4,
leftMargin=1.8*cm, rightMargin=1.8*cm,
topMargin=2*cm, bottomMargin=2*cm,
title="Pathophysiology of Fever – Viva Study Guide",
author="1st Year Resident, Physiology"
)
# ── Styles ────────────────────────────────────────────────────────────────────
base = getSampleStyleSheet()
def S(name, **kw):
return ParagraphStyle(name, **kw)
cover_title = S("CoverTitle",
fontSize=28, textColor=WHITE, fontName="Helvetica-Bold",
alignment=TA_CENTER, spaceAfter=8, leading=34)
cover_sub = S("CoverSub",
fontSize=13, textColor=colors.HexColor("#cce8f4"), fontName="Helvetica",
alignment=TA_CENTER, spaceAfter=6, leading=18)
cover_date = S("CoverDate",
fontSize=10, textColor=colors.HexColor("#aad4e8"), fontName="Helvetica-Oblique",
alignment=TA_CENTER)
section_head = S("SectionHead",
fontSize=13, textColor=WHITE, fontName="Helvetica-Bold",
alignment=TA_LEFT, spaceBefore=14, spaceAfter=6, leading=17,
backColor=TEAL, borderPadding=(5,8,5,8))
q_label = S("QLabel",
fontSize=10, textColor=NAVY, fontName="Helvetica-Bold",
spaceBefore=10, spaceAfter=2, leading=14)
q_text = S("QText",
fontSize=10.5, textColor=colors.HexColor("#1a1a2e"), fontName="Helvetica-Bold",
spaceBefore=0, spaceAfter=4, leading=14, leftIndent=10)
a_label = S("ALabel",
fontSize=9.5, textColor=TEAL, fontName="Helvetica-Bold",
spaceBefore=2, spaceAfter=1)
a_text = S("AText",
fontSize=10, textColor=colors.HexColor("#1c1c1c"), fontName="Helvetica",
spaceBefore=0, spaceAfter=6, leading=14, leftIndent=10,
alignment=TA_JUSTIFY)
tip_style = S("Tip",
fontSize=9.5, textColor=colors.HexColor("#7d4600"), fontName="Helvetica-Oblique",
spaceBefore=2, spaceAfter=4, leading=13, leftIndent=10,
backColor=WARN_BG, borderPadding=4)
note_style = S("Note",
fontSize=9.5, textColor=colors.HexColor("#145a32"), fontName="Helvetica-Oblique",
spaceBefore=2, spaceAfter=6, leading=13, leftIndent=10,
backColor=Q_BG, borderPadding=4)
bullet_style = S("Bullet",
fontSize=10, textColor=colors.HexColor("#1c1c1c"), fontName="Helvetica",
spaceBefore=1, spaceAfter=1, leading=13, leftIndent=20,
bulletIndent=10, bulletText="\u2022")
toc_style = S("TOC",
fontSize=11, textColor=NAVY, fontName="Helvetica",
spaceBefore=4, spaceAfter=4, leading=16)
footer_style = S("Footer",
fontSize=8, textColor=colors.grey, fontName="Helvetica-Oblique",
alignment=TA_CENTER)
# ── Page template with header/footer ─────────────────────────────────────────
def on_page(canvas, doc):
canvas.saveState()
w, h = A4
# top bar
canvas.setFillColor(NAVY)
canvas.rect(0, h-1.2*cm, w, 1.2*cm, fill=1, stroke=0)
canvas.setFont("Helvetica-Bold", 8)
canvas.setFillColor(WHITE)
canvas.drawString(1.8*cm, h-0.8*cm, "PATHOPHYSIOLOGY OF FEVER | VIVA STUDY GUIDE")
canvas.drawRightString(w-1.8*cm, h-0.8*cm, "1st Year Resident – Physiology")
# bottom bar
canvas.setFillColor(TEAL)
canvas.rect(0, 0, w, 1*cm, fill=1, stroke=0)
canvas.setFont("Helvetica", 8)
canvas.setFillColor(WHITE)
canvas.drawCentredString(w/2, 0.35*cm, f"Page {doc.page}")
canvas.restoreState()
def on_first_page(canvas, doc):
pass # cover page – no header/footer
# ── Helper builders ───────────────────────────────────────────────────────────
def section(title):
return [
Spacer(1, 0.3*cm),
Table([[Paragraph(title, section_head)]],
colWidths=[doc.width],
style=TableStyle([
("BACKGROUND", (0,0), (-1,-1), TEAL),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING", (0,0), (-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 10),
])),
Spacer(1, 0.15*cm),
]
def qa(num, question, answer_parts, tip=None, note=None):
"""answer_parts = list of strings; each becomes a paragraph."""
elems = []
# Question box
q_data = [[
Paragraph(f"Q{num}", q_label),
Paragraph(question, q_text)
]]
qt = Table(q_data, colWidths=[1.2*cm, doc.width-1.2*cm],
style=TableStyle([
("BACKGROUND", (0,0), (-1,-1), LIGHT_BG),
("TOPPADDING", (0,0), (-1,-1), 6),
("BOTTOMPADDING", (0,0), (-1,-1), 6),
("LEFTPADDING", (0,0), (-1,-1), 6),
("VALIGN", (0,0), (-1,-1), "TOP"),
("LINEBELOW", (0,0), (-1,-1), 0.5, TEAL),
]))
elems.append(qt)
elems.append(Paragraph("A:", a_label))
for part in answer_parts:
elems.append(Paragraph(part, a_text))
if tip:
elems.append(Paragraph(f"Examiner tip: {tip}", tip_style))
if note:
elems.append(Paragraph(f"Key fact: {note}", note_style))
elems.append(HRFlowable(width="100%", thickness=0.3, color=colors.lightgrey,
spaceAfter=4))
return KeepTogether(elems)
def bullet(text):
return Paragraph(text, bullet_style)
# ══════════════════════════════════════════════════════════════════════════════
# CONTENT
# ══════════════════════════════════════════════════════════════════════════════
story = []
# ── COVER PAGE ────────────────────────────────────────────────────────────────
cover_table_data = [[
Paragraph("PATHOPHYSIOLOGY OF FEVER", cover_title),
]]
cover_bg = Table(
[
[Spacer(1, 2*cm)],
[Paragraph("PATHOPHYSIOLOGY OF FEVER", cover_title)],
[Spacer(1, 0.4*cm)],
[Paragraph("Viva Q&A Study Guide", cover_sub)],
[Spacer(1, 0.2*cm)],
[Paragraph("For 1st Year Physiology Residents", cover_sub)],
[Spacer(1, 0.6*cm)],
[HRFlowable(width="60%", thickness=1.5, color=AMBER, spaceAfter=8)],
[Spacer(1, 0.4*cm)],
[Paragraph("18 High-Yield Questions | All Categories Covered", cover_sub)],
[Spacer(1, 0.3*cm)],
[Paragraph("Basic \u2022 Mechanism-Based \u2022 Applied \u2022 Tricky Examiner Favourites", cover_sub)],
[Spacer(1, 2*cm)],
[Paragraph("May 2026", cover_date)],
[Spacer(1, 3*cm)],
],
colWidths=[doc.width + 3.6*cm],
style=TableStyle([
("BACKGROUND", (0,0), (-1,-1), NAVY),
("ALIGN", (0,0), (-1,-1), "CENTER"),
("TOPPADDING", (0,0), (-1,-1), 0),
("BOTTOMPADDING", (0,0), (-1,-1), 0),
("LEFTPADDING", (0,0), (-1,-1), 0),
("RIGHTPADDING", (0,0), (-1,-1), 0),
])
)
story.append(cover_bg)
story.append(PageBreak())
# ── HOW TO USE ────────────────────────────────────────────────────────────────
story += section("HOW TO USE THIS GUIDE")
story.append(Paragraph(
"This guide contains 18 viva questions across 4 difficulty tiers. "
"Cover the answer with your hand or a sheet of paper, attempt the question, "
"then reveal the answer. Pay special attention to <b>Examiner tips</b> (orange boxes) "
"and <b>Key facts</b> (green boxes) - these highlight what separates a good answer "
"from an excellent one.",
a_text))
story.append(Spacer(1, 0.2*cm))
tier_data = [
["Tier", "Questions", "Focus"],
["BASIC / OPENING", "Q1 - Q3", "Definitions, normal values, terminology"],
["MECHANISM-BASED", "Q4 - Q8", "Biochemical pathways, drug targets"],
["TRICKY / APPLIED", "Q9 - Q14", "Clinical scenarios, nuanced reasoning"],
["EXAMINER FAVOURITES", "Q15 - Q18", "Classic traps, comparison questions"],
]
tier_table = Table(tier_data, colWidths=[5*cm, 3*cm, doc.width-8*cm],
style=TableStyle([
("BACKGROUND", (0,0), (-1,0), NAVY),
("TEXTCOLOR", (0,0), (-1,0), WHITE),
("FONTNAME", (0,0), (-1,0), "Helvetica-Bold"),
("FONTSIZE", (0,0), (-1,-1), 9.5),
("BACKGROUND", (0,1), (-1,1), colors.HexColor("#e8f4ea")),
("BACKGROUND", (0,2), (-1,2), LIGHT_BG),
("BACKGROUND", (0,3), (-1,3), colors.HexColor("#fef9e7")),
("BACKGROUND", (0,4), (-1,4), colors.HexColor("#fde8e8")),
("ROWBACKGROUNDS", (0,1), (-1,-1), [colors.HexColor("#f0f6fb"), WHITE]),
("GRID", (0,0), (-1,-1), 0.4, colors.lightgrey),
("TOPPADDING", (0,0), (-1,-1), 5),
("BOTTOMPADDING", (0,0), (-1,-1), 5),
("LEFTPADDING", (0,0), (-1,-1), 8),
]))
story.append(tier_table)
story.append(PageBreak())
# ══════════════════════════════════════════════════════════════════════════════
# SECTION 1 – BASIC
# ══════════════════════════════════════════════════════════════════════════════
story += section("TIER 1 – BASIC / OPENING QUESTIONS")
story.append(qa(1,
"What is the normal body temperature and how is it regulated?",
[
"Normal oral temperature is <b>36.0°C – 37.8°C (96.8°F – 100.0°F)</b>. "
"It follows a <b>circadian rhythm</b> – lowest at 6 AM, highest at 4–6 PM.",
"Regulation is by the <b>preoptic anterior hypothalamus</b>, which acts as the "
"body's thermostat. Multiple temperature sensors provide feedback via neural "
"pathways. The hypothalamus coordinates heat gain/loss through: "
"(1) vasomotor changes, (2) shivering, (3) metabolic heat production, "
"and (4) behavioral changes.",
],
note="The preoptic anterior hypothalamus is the key thermoregulatory centre – "
"always name it specifically, not just 'the hypothalamus'."
))
story.append(qa(2,
"What temperature defines fever? What is the CDC definition?",
[
"There is <b>no universal consensus</b>. The <b>CDC</b> defines fever as a core "
"temperature <b>>38.0°C (100.4°F)</b> in the absence of fever-reducing "
"medication.",
"Most clinicians consider <b>>38.3°C (100.9°F)</b> a significant fever. "
"Temperatures <b>>41°C (105.8°F)</b> are usually due to <b>hyperthermia</b> "
"rather than true fever.",
],
tip="Examiners love asking about the 38°C vs 38.3°C distinction – mention both."
))
story.append(qa(3,
"What is a pyrogen? Give one exogenous and one endogenous example.",
[
"A <b>pyrogen</b> is any substance that induces fever (from Greek: pyro = fire, "
"gen = producing).",
"<b>Exogenous pyrogen:</b> LPS (lipopolysaccharide / endotoxin) from the outer "
"membrane of gram-negative bacteria – the classic example.",
"<b>Endogenous pyrogen:</b> IL-1β (Interleukin-1 beta) – a cytokine released by "
"activated macrophages and monocytes. TNF-α, IL-6, and IFN-γ are also "
"endogenous pyrogens.",
],
note="Exogenous pyrogens do NOT act directly on the hypothalamus – they first trigger "
"endogenous pyrogen release from immune cells."
))
story.append(PageBreak())
# ══════════════════════════════════════════════════════════════════════════════
# SECTION 2 – MECHANISM
# ══════════════════════════════════════════════════════════════════════════════
story += section("TIER 2 – MECHANISM-BASED QUESTIONS")
story.append(qa(4,
"What is the single most important mediator in the final pathway of fever generation?",
[
"<b>Prostaglandin E2 (PGE2)</b> is the final common mediator. All pyrogenic "
"signals – whether exogenous (LPS via TLR-4) or endogenous (IL-1β, TNF-α, "
"IL-6) – converge on the hypothalamus through PGE2.",
"PGE2 acts on <b>EP1 and EP3 receptors</b> in the preoptic area, raising the "
"thermoregulatory set point. This is why blocking PGE2 synthesis (NSAIDs) "
"reduces fever regardless of the cause.",
],
tip="If asked 'what is the final common pathway of fever?' – answer: PGE2 acting "
"on hypothalamic EP receptors."
))
story.append(qa(5,
"Which enzyme is the key NSAID target in fever? Difference between COX-1 and COX-2?",
[
"The key enzyme is <b>Cyclooxygenase-2 (COX-2)</b> – it is <b>inducible</b>, "
"upregulated in the hypothalamus in response to IL-1β and TNF-α.",
"<b>COX-1</b> = constitutive ('housekeeping') isoform. Found in most tissues. "
"Protects gastric mucosa, aids platelet aggregation.",
"<b>COX-2</b> = inducible isoform. Responsible for pathological PGE2 in fever "
"and inflammation.",
"Traditional NSAIDs (ibuprofen, aspirin) inhibit <b>both</b>. Selective COX-2 "
"inhibitors (celecoxib) spare COX-1, reducing GI side effects.",
],
note="COX-2 is your target for antipyresis; COX-1 inhibition is responsible for "
"GI and platelet side effects."
))
story.append(qa(6,
"Mechanistic difference between aspirin and ibuprofen as antipyretics?",
[
"<b>Aspirin:</b> <b>Irreversible</b> acetylation of the serine residue in the "
"active site of both COX-1 and COX-2. New enzyme must be synthesised. "
"In platelets (no nucleus), effect is permanent for the platelet's lifetime "
"(~10 days).",
"<b>Ibuprofen:</b> <b>Reversible, competitive</b> inhibition of both COX "
"isoforms. Effect wears off as drug is cleared.",
"Both reduce fever by the same endpoint: ↓ PGE2 → set point normalises → "
"defervescence.",
],
tip="Aspirin's irreversible action on platelets is why it is used for "
"antiplatelet therapy but avoided in children (Reye syndrome risk)."
))
story.append(qa(7,
"Why does paracetamol reduce fever but has no significant anti-inflammatory action?",
[
"Paracetamol inhibits COX enzymes <b>centrally (in the CNS)</b> but has very "
"weak peripheral inhibition.",
"In the lipid-rich, <b>low-peroxide</b> CNS environment, it can inhibit COX "
"effectively. In inflamed peripheral tissues with <b>high peroxide "
"concentrations</b> (from neutrophil activity), paracetamol is oxidised and "
"rendered inactive.",
"Result: ↓ hypothalamic PGE2 (→ <b>antipyretic</b>) but no suppression of "
"peripheral prostaglandin synthesis (→ <b>no anti-inflammatory effect</b>).",
],
note="This is why paracetamol is NOT classed as an NSAID – it has no significant "
"peripheral anti-inflammatory action."
))
story.append(qa(8,
"Step at which LPS (endotoxin) first acts to generate fever?",
[
"1. LPS binds to <b>CD14</b> (co-receptor on macrophages) in complex with "
"<b>LPS-binding protein (LBP)</b>.",
"2. This complex signals through <b>TLR-4 (Toll-Like Receptor 4)</b> on the "
"macrophage surface.",
"3. TLR-4 signals via <b>MyD88</b> and <b>TRIF</b> adaptor proteins → activates "
"<b>NF-κB</b>.",
"4. NF-κB drives transcription of pyrogenic cytokines (IL-1β, TNF-α, IL-6) "
"AND directly induces <b>COX-2</b> → PGE2 synthesis → fever.",
],
tip="Remember the sequence: LPS → LBP+CD14 → TLR-4 → MyD88/TRIF → NF-κB → "
"cytokines + COX-2 → PGE2 → hypothalamus."
))
story.append(PageBreak())
# ══════════════════════════════════════════════════════════════════════════════
# SECTION 3 – TRICKY / APPLIED
# ══════════════════════════════════════════════════════════════════════════════
story += section("TIER 3 – TRICKY / APPLIED QUESTIONS")
story.append(qa(9,
"A patient has 42°C after working in the heat. IV paracetamol does not help. Why?",
[
"This is <b>hyperthermia</b>, not fever. In hyperthermia, the hypothalamic set "
"point is <b>normal</b> – the problem is inability to dissipate excess heat. "
"No PGE2-mediated set point elevation is occurring.",
"Antipyretics (NSAIDs, paracetamol) work by <b>lowering PGE2</b> → reducing "
"set point. Since the set point is already normal in hyperthermia, they have "
"<b>no effect</b>.",
"<b>Management:</b> Physical cooling – ice packs, cooling blankets, cool IV "
"fluids, move to cool environment. In malignant hyperthermia: dantrolene "
"(blocks ryanodine receptor → stops uncontrolled Ca2+ release).",
],
tip="The examiner wants you to state clearly: 'antipyretics are ineffective in "
"hyperthermia because they target PGE2, which is not elevated here.'"
))
story.append(qa(10,
"If both fever and hyperthermia raise temperature, why is hyperthermia more dangerous?",
[
"<b>1. No ceiling mechanism:</b> In fever, endogenous antipyretics (AVP, α-MSH, "
"IL-10, glucocorticoids) and the fixed dose of pyrogen limit the rise – usually "
"not beyond 41°C. In hyperthermia, there is no regulatory ceiling; "
"temperatures can exceed 42–43°C.",
"<b>2. No adaptive response:</b> In fever, the body 'wants' the higher "
"temperature. In hyperthermia, the hypothalamus is desperately trying to cool "
"down but cannot. Temperatures above 41–42°C cause <b>direct protein "
"denaturation</b>, enzyme dysfunction, and multi-organ failure.",
],
note="Fever has a built-in 'thermostat with a new setting'; hyperthermia is a "
"'thermostat overwhelmed by heat load'."
))
story.append(qa(11,
"What are endogenous antipyretics? Why doesn't fever go on indefinitely?",
[
"The body has built-in negative feedback to limit fever:",
"• <b>Arginine vasopressin (AVP)</b> – released from hypothalamus; acts "
"centrally to limit fever",
"• <b>α-Melanocyte-stimulating hormone (α-MSH)</b> – potent antipyretic and "
"anti-inflammatory neuropeptide",
"• <b>IL-10</b> – anti-inflammatory cytokine; inhibits macrophage activation "
"and cytokine production",
"• <b>Glucocorticoids</b> – cortisol suppresses cytokine production and "
"PGE2 synthesis (stress response)",
"As fever rises, these signals rise proportionately, preventing runaway "
"temperature elevation.",
],
tip="Naming even 2 endogenous antipyretics will impress the examiner – most "
"candidates only say 'the body regulates itself'."
))
story.append(qa(12,
"Why can febrile seizures occur at 38–39°C when fever rarely exceeds 41°C?",
[
"These are <b>independent variables</b>:",
"<b>Upper limit of fever</b> is controlled by endogenous antipyretics (AVP, "
"α-MSH, IL-10) which activate as temperature rises.",
"<b>Febrile seizures</b> depend not on absolute temperature but on the "
"<b>rate of temperature rise</b>. A rapid rise from 37°C to 38.5°C can trigger "
"a seizure as readily as a higher temperature.",
"The immature, incompletely myelinated brain of young children has a "
"<b>lower seizure threshold</b> and is highly sensitive to rapid temperature "
"changes affecting ion channel kinetics and neuronal excitability.",
],
note="Rate of rise > absolute height in febrile seizure pathogenesis."
))
story.append(qa(13,
"Child with 3 febrile seizures by age 4. Risk of epilepsy? Factors worsening prognosis?",
[
"Baseline risk of later epilepsy after febrile seizures: <b>~7% by age 25</b> "
"(vs. ~1% general population).",
"Risk increases with <b>complex features</b>: (1) duration >15 min, "
"(2) focal features, (3) >1 seizure in 24 hours.",
"• 1 complex feature: <b>6–8%</b>",
"• 2 complex features: <b>17–22%</b>",
"• All 3 complex features: <b>~49%</b>",
"<b>Additional poor prognostic factors:</b> pre-existing neurodevelopmental "
"abnormality, family history of epilepsy, febrile status epilepticus "
"(hippocampal injury → sclerosis → temporal lobe epilepsy).",
],
tip="Quote the Annegers et al. (1987) study figures – examiners love specific numbers."
))
story.append(qa(14,
"Parents ask: should we start phenobarbital after child's first simple febrile seizure?",
[
"<b>No</b> – advise against chronic prophylaxis.",
"Evidence (Farwell et al., 1990): phenobarbital can reduce <i>recurrence</i> "
"of febrile seizures but does <b>NOT reduce risk of later epilepsy</b>, and "
"carries <b>significant cognitive side effects</b> in developing children "
"(impaired IQ, behavioral problems).",
"Benefits do not outweigh risks for simple febrile seizures.",
"<b>Preferred approach for high-risk children:</b> <b>Intermittent rectal "
"diazepam</b> administered at onset of fever – prevents seizures without "
"daily drug toxicity.",
],
note="Simple febrile seizures – benign, no chronic prophylaxis. "
"Complex febrile seizures – evaluate further, consider intermittent diazepam."
))
story.append(PageBreak())
# ══════════════════════════════════════════════════════════════════════════════
# SECTION 4 – EXAMINER FAVOURITES
# ══════════════════════════════════════════════════════════════════════════════
story += section("TIER 4 – CLASSIC TRAPS / EXAMINER FAVOURITES")
story.append(qa(15,
"A patient with severe sepsis does NOT develop fever. Is this a good sign?",
[
"<b>No – it is a bad sign.</b>",
"Fever is a <b>protective host response</b>. Patients with severe bacterial "
"infection who fail to mount fever have <b>higher morbidity and mortality</b> "
"(Goldman-Cecil Medicine).",
"Absence of fever in sepsis usually indicates:",
"• Overwhelming infection with immune exhaustion",
"• Elderly or immunocompromised patient (blunted cytokine response)",
"• Antipyretic medications masking fever",
"• Often heralds development of <b>septic shock</b> and worse outcomes.",
],
tip="This is a classic reversal question. The expected wrong answer is 'good sign "
"because no fever = less inflammation'. Always remember: no fever in severe "
"infection = poor prognosis."
))
story.append(qa(16,
"What is temperature-pulse dissociation? In which fevers is it classically seen?",
[
"Normally, heart rate rises <b>2–5 beats/min per 1°F</b> increase in temperature "
"(Liebermeister's rule). <b>Temperature-pulse dissociation (relative "
"bradycardia)</b> = absence of this expected tachycardia despite significant "
"fever.",
"<b>Classic causes (mnemonic: T-BIRD-L):</b>",
"• <b>T</b>yphoid fever (Salmonella typhi)",
"• <b>B</b>abesiosis",
"• <b>I</b>nfluenza (relative – less dramatic)",
"• <b>R</b>ickettsiosis",
"• <b>D</b>engue fever",
"• <b>L</b>eptospirosis / <b>L</b>egionellosis",
],
note="Mechanism: direct autonomic effects of the pathogen or toxin on cardiac "
"conduction, overriding the normal sympathetic tachycardia."
))
story.append(qa(17,
"How does iron sequestration during fever help fight infection? Is it always beneficial?",
[
"<b>Mechanism of benefit:</b> During acute-phase response, <b>hepcidin</b> "
"(stimulated by IL-6) promotes iron sequestration in macrophages and reduces "
"serum iron. Many pathogens require iron for growth (siderophore production), "
"so low serum iron is <b>bacteriostatic</b>. Lactoferrin and ferritin also "
"bind free iron.",
"<b>Not always beneficial:</b> In <b>anaemia of chronic disease / "
"inflammation</b>, prolonged iron sequestration causes a hypochromic "
"normocytic/microcytic anaemia that does <b>not</b> respond to iron "
"supplementation, because the problem is sequestration, not deficiency.",
],
tip="This question links fever physiology to clinical haematology – a cross-discipline "
"connection that impresses examiners."
))
story.append(qa(18,
"For every 1°C rise in body temperature, what happens to the basal metabolic rate?",
[
"BMR increases by approximately <b>10–13% per 1°C rise</b> "
"(~7% per 1°F rise).",
"<b>Metabolic consequences of sustained fever:</b>",
"• <b>↑ O2 consumption</b> and ↑ CO2 production",
"• <b>↑ Protein catabolism</b> → negative nitrogen balance, muscle wasting",
"• <b>↑ Gluconeogenesis</b> (driven by IL-1, TNF-α) to meet fuel demands",
"• <b>↑ Lipolysis</b> and brown fat catabolism",
"• <b>↑ Heart rate</b> (~2–5 beats/min per 1°F)",
"• <b>↑ Respiratory rate</b> (driven by ↑ O2 demand)",
"• <b>Water and electrolyte loss</b> via sweating during defervescence",
"• <b>↑ Acute-phase proteins</b> (CRP, fibrinogen, SAA) – liver response to IL-6",
"• <b>↓ Albumin, transferrin</b> (negative acute-phase reactants)",
],
note="A prolonged fever of 40°C increases daily caloric requirement by ~40%. "
"This is why nutritional support matters in febrile ICU patients."
))
story.append(PageBreak())
# ══════════════════════════════════════════════════════════════════════════════
# QUICK REFERENCE SUMMARY TABLE
# ══════════════════════════════════════════════════════════════════════════════
story += section("QUICK REFERENCE: FEVER VS HYPERTHERMIA")
fvh_data = [
["Feature", "FEVER", "HYPERTHERMIA"],
["Set point", "RAISED (by PGE2)", "Normal"],
["Mechanism", "Regulated (pyrogen cascade)", "Heat gain > dissipation"],
["Antipyretics", "EFFECTIVE", "Ineffective"],
["Shivering/chills", "Yes (rising phase)", "No"],
["Max temp", "Usually <41°C", "Can exceed 42–43°C"],
["Sweating initially", "No (vasoconstriction)", "Yes (trying to cool)"],
["Treatment", "Antipyretics + treat cause", "Physical cooling"],
["Examples", "Infection, malignancy, autoimmune", "Heat stroke, MH, NMS"],
]
fvh_table = Table(fvh_data,
colWidths=[4.5*cm, 6*cm, 6*cm],
style=TableStyle([
("BACKGROUND", (0,0), (-1,0), NAVY),
("TEXTCOLOR", (0,0), (-1,0), WHITE),
("FONTNAME", (0,0), (-1,0), "Helvetica-Bold"),
("FONTNAME", (0,0), (0,-1), "Helvetica-Bold"),
("BACKGROUND", (1,0), (1,0), TEAL),
("BACKGROUND", (2,0), (2,0), colors.HexColor("#c0392b")),
("FONTSIZE", (0,0), (-1,-1), 9),
("ROWBACKGROUNDS", (0,1), (-1,-1), [LIGHT_BG, WHITE]),
("GRID", (0,0), (-1,-1), 0.4, colors.grey),
("TOPPADDING", (0,0), (-1,-1), 5),
("BOTTOMPADDING", (0,0), (-1,-1), 5),
("LEFTPADDING", (0,0), (-1,-1), 7),
("ALIGN", (0,0), (-1,-1), "LEFT"),
("VALIGN", (0,0), (-1,-1), "MIDDLE"),
]))
story.append(fvh_table)
story.append(Spacer(1, 0.5*cm))
# ── Pyrogen summary ───────────────────────────────────────────────────────────
story += section("QUICK REFERENCE: PYROGENS AT A GLANCE")
pyr_data = [
["EXOGENOUS PYROGENS", "ENDOGENOUS PYROGENS (Cytokines)"],
["LPS/Endotoxin (gram-negative bacteria)", "IL-1β (classic endogenous pyrogen)"],
["Peptidoglycan / Lipoteichoic acid (gram+)", "TNF-α"],
["Superantigens (TSST-1, staph enterotoxins)", "IL-6 (also drives acute-phase response)"],
["Viral dsRNA, capsid proteins", "IFN-γ (T cells, NK cells)"],
["Fungal mannans, zymosan", ""],
["Blood products, some drugs", ""],
["Action: TLR-4 → NF-κB → cytokines", "Action: → COX-2 → PGE2 → hypothalamus"],
]
pyr_table = Table(pyr_data,
colWidths=[doc.width/2, doc.width/2],
style=TableStyle([
("BACKGROUND", (0,0), (-1,0), TEAL),
("TEXTCOLOR", (0,0), (-1,0), WHITE),
("FONTNAME", (0,0), (-1,0), "Helvetica-Bold"),
("FONTSIZE", (0,0), (-1,-1), 9),
("ROWBACKGROUNDS", (0,1), (-1,-1), [LIGHT_BG, WHITE]),
("GRID", (0,0), (-1,-1), 0.4, colors.grey),
("TOPPADDING", (0,0), (-1,-1), 5),
("BOTTOMPADDING", (0,0), (-1,-1), 5),
("LEFTPADDING", (0,0), (-1,-1), 7),
("VALIGN", (0,0), (-1,-1), "MIDDLE"),
("BACKGROUND", (0,7), (-1,7), colors.HexColor("#e8f4ea")),
("FONTNAME", (0,7), (-1,7), "Helvetica-BoldOblique"),
]))
story.append(pyr_table)
story.append(Spacer(1, 0.5*cm))
# ── Fever cascade ─────────────────────────────────────────────────────────────
story += section("FEVER CASCADE – AT A GLANCE")
cascade_text = (
"<b>Infection / Inflammation / Neoplasm</b> → Immune cells (macrophages, monocytes) "
"→ Exogenous pyrogens trigger endogenous pyrogens (IL-1β, TNF-α, IL-6, IFN-γ) "
"→ Preoptic anterior hypothalamus → ↑ COX-2 → ↑ PGE2 (EP1 / EP3 receptors) "
"→ <b>RAISED SET POINT</b> → Vasoconstriction + Shivering (<b>CHILLS – rising phase</b>) "
"→ Core temp reaches new set point (<b>FEVER maintained</b>) "
"→ Cause resolves / NSAIDs → ↓ PGE2 → set point normalises "
"→ Vasodilation + Sweating (<b>DEFERVESCENCE</b>)"
)
cascade_table = Table([[Paragraph(cascade_text, a_text)]],
colWidths=[doc.width],
style=TableStyle([
("BACKGROUND", (0,0), (-1,-1), colors.HexColor("#eaf4fb")),
("GRID", (0,0), (-1,-1), 0.5, TEAL),
("TOPPADDING", (0,0), (-1,-1), 8),
("BOTTOMPADDING", (0,0), (-1,-1), 8),
("LEFTPADDING", (0,0), (-1,-1), 10),
]))
story.append(cascade_table)
story.append(Spacer(1, 0.4*cm))
# ── References ────────────────────────────────────────────────────────────────
story += section("SOURCES")
refs = [
"Rosen's Emergency Medicine – Concepts and Clinical Practice (Pathophysiology of Fever, p. 123)",
"Robbins, Cotran & Kumar – Pathologic Basis of Disease (Systemic Effects of Inflammation)",
"Goldman-Cecil Medicine, International Edition (Pathobiology of Fever)",
"Janeway's Immunobiology, 10e (Acute-Phase Response, Section 3-21)",
"Goodman & Gilman's Pharmacological Basis of Therapeutics (Febrile Convulsions)",
"Bradley and Daroff's Neurology in Clinical Practice (Febrile Seizures)",
"Katzung's Basic and Clinical Pharmacology, 16th Ed. (Fever, Chapter on NSAIDs)",
]
for r in refs:
story.append(Paragraph(f"\u2022 {r}", bullet_style))
story.append(Spacer(1, 0.5*cm))
story.append(Paragraph(
"Study guide compiled for Physiology Seminar – Pathophysiology of Fever | May 2026",
footer_style))
# ── BUILD ─────────────────────────────────────────────────────────────────────
doc.build(story,
onFirstPage=on_first_page,
onLaterPages=on_page)
print(f"PDF created: {OUTPUT}")
cd /home/daytona/workspace/fever-study-guide && python3 generate_pdf.py
ls -lh /home/daytona/workspace/fever-study-guide/Fever_Viva_Study_Guide.pdf
Fever Viva Study Guide
PDF Document · PDF
| Section | Content |
|---|---|
| Cover page | Title, tier breakdown, date |
| How to Use | Tier overview table with focus areas |
| Tier 1 – Basic | Q1–Q3: Normal temp, fever definition, pyrogens |
| Tier 2 – Mechanism | Q4–Q8: PGE2, COX-1 vs COX-2, aspirin vs ibuprofen, paracetamol, LPS cascade |
| Tier 3 – Applied | Q9–Q14: Hyperthermia scenario, endogenous antipyretics, febrile seizures, prophylaxis |
| Tier 4 – Examiner Traps | Q15–Q18: No fever in sepsis, pulse-temp dissociation, iron sequestration, metabolic effects |
| Quick Reference Tables | Fever vs hyperthermia side-by-side, pyrogens at a glance |
| Fever Cascade | Full pathway from trigger to defervescence |
| Sources | All 7 textbooks cited |