nephrotic syndrome
Reading File
Finding Sources
Searching PubMed
"nephrotic syndrome"[MeSH Terms] AND management
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
I now have comprehensive material from multiple authoritative sources. Let me compile the full answer.
Nephrotic Syndrome
Definition
Nephrotic syndrome is a clinical complex defined by proteinuria >3.5 g/day (in adults), hypoalbuminemia (serum albumin <3 g/dL), generalized edema, and hyperlipidemia with lipiduria. It results from any glomerular disorder that disrupts the normal charge and size selectivity of the glomerular filtration barrier, allowing large molecules such as albumin to escape into the urine.
Pathophysiology
The glomerular filtration barrier is maintained by three components working together: the glomerular basement membrane (GBM), endothelial cells, and epithelial cells (podocytes). When these are injured - by immune dysregulation, circulating permeability factors, or systemic disease - glomerular permeability to proteins increases.
The Tetrad of Features
| Feature | Mechanism |
|---|---|
| Proteinuria | Impaired glomerular size/charge selectivity; albumin and other proteins escape |
| Hypoalbuminemia | Urinary losses exceed hepatic synthesis; compensatory increase in synthesis is blunted |
| Hyperlipidemia/Lipiduria | Increased hepatic lipoprotein synthesis, impaired lipid catabolism, abnormal transport; lipiduria from increased GBM permeability to lipoproteins |
| Edema | Low plasma oncotic pressure drives fluid into the interstitium (underfill) + primary renal Na+ retention (overfill) |
Mechanisms of Nephrotic Edema
Two distinct and often coexisting mechanisms produce edema:
- Underfill: Hypoalbuminemia lowers plasma oncotic pressure → fluid shifts to interstitium → reduced plasma volume → RAAS activation → aldosterone-driven Na+ retention → worsens edema
- Overfill (more common): Primary defect in distal nephron Na+ excretion (likely ENaC activation by proteolytic enzymes in tubular fluid) → Na+ and water retention → expanded plasma volume, suppressed RAAS
Minimal change disease typically follows the underfill pattern with contracted plasma volume and activated RAAS; most other causes follow the overfill pattern.
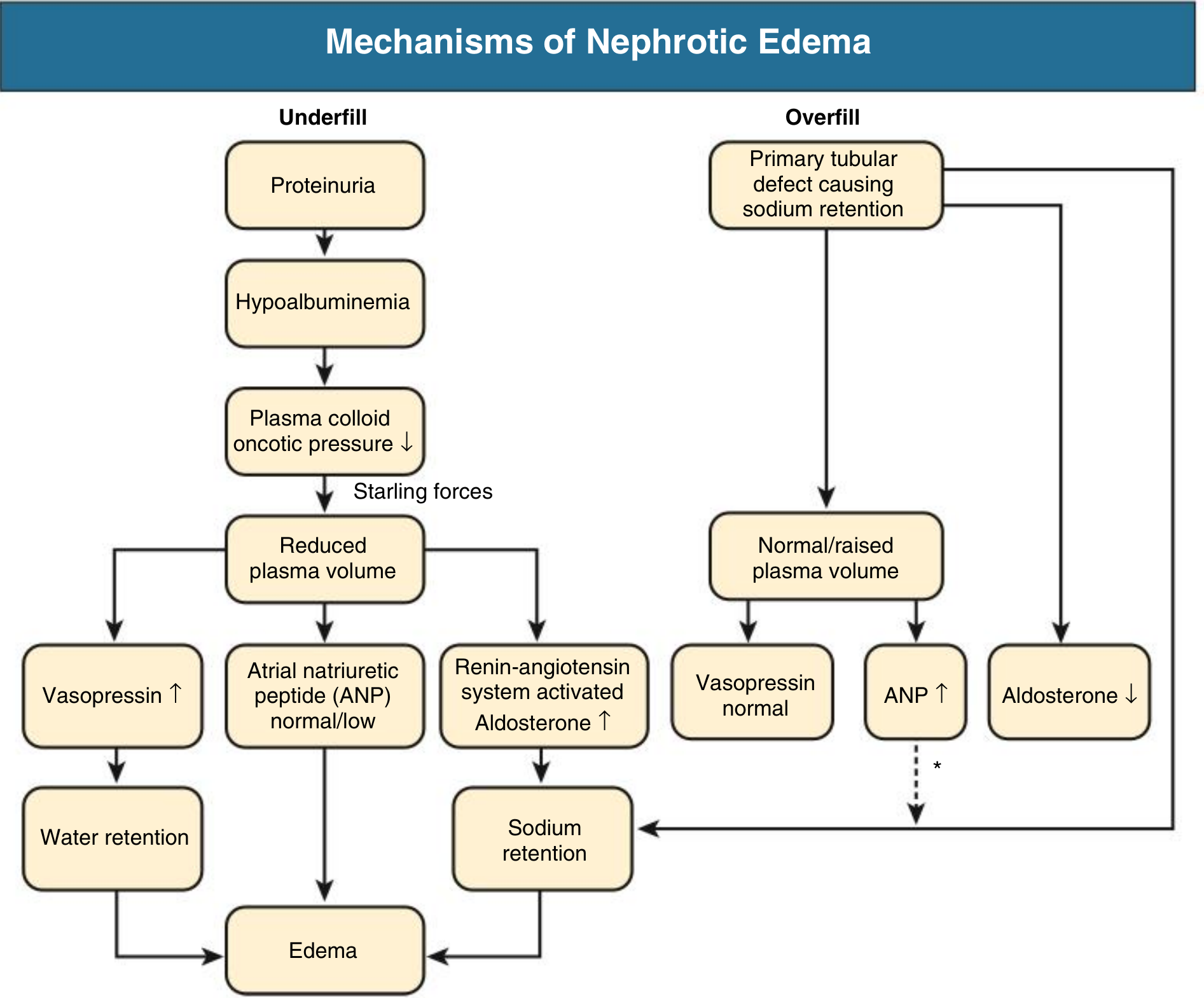
Causes
Primary (Idiopathic) Glomerular Diseases
| Disease | Key Features | Age Group |
|---|---|---|
| Minimal Change Disease (MCD) | Normal LM; podocyte foot process effacement on EM; responds to steroids (>80% remission); relapses common | Most common in children; up to 15% of adult cases |
| Focal Segmental Glomerulosclerosis (FSGS) | Focal, segmental sclerosis of glomeruli; higher prevalence in African Americans; often steroid resistant | Adolescents/adults |
| Membranous Nephropathy (MN) | Subepithelial immune deposits; PLA2R antibody in primary form; most common adult primary nephrotic syndrome | Adults >40 yr |
| Membranoproliferative GN (MPGN) | Mesangial expansion + double contour ("tram-track") GBM | Any age |
| IgA Nephropathy | Mainly nephritic, but ~10-15% can present with nephrotic range proteinuria | Young adults |
Secondary Causes
- Diabetes mellitus - Kimmelstiel-Wilson nodular glomerulosclerosis; most common secondary cause
- Lupus (SLE) - can mirror membranous or other patterns (lupus nephritis class V for nephrotic)
- Preeclampsia - nephrotic-range proteinuria + hypertension in pregnancy
- Infections - Hepatitis B (membranous pattern), hepatitis C, HIV, malaria, infective endocarditis
- Malignancy - Hodgkin lymphoma (classically associated with MCD), carcinomas (membranous)
- Drugs - Gold, penicillamine (membranous), heroin, NSAIDs, captopril
- Amyloidosis - AL or AA amyloid deposited in glomeruli; Congo red staining with apple-green birefringence
Clinical Features
Symptoms
- Foamy urine (frothy due to proteinuria)
- Peripheral edema - often worse in the morning, periorbital edema (especially in children); may progress to anasarca (generalized severe edema including ascites, scrotal/vulvar edema, pleural effusions)
- Fatigue, dyspnea, orthopnea
- Nausea/vomiting (from bowel wall edema or ascites)
Signs
- Hypertension, pitting edema
- Ascites, pleural effusions
- Muehrcke's lines - white transverse bands on nails (hypoalbuminemia sign)
- Eruptive xanthomata, xanthelasma (hyperlipidemia)
Laboratory Findings
- Proteinuria >3.5 g/day (or spot urine protein:creatinine ratio >3.5)
- Serum albumin <2.5-3 g/dL
- Total cholesterol usually >180 mg/dL; raised LDL and triglycerides
- Lipiduria on urinalysis (fatty casts, oval fat bodies)
- Serum creatinine variable
Complications
1. Thromboembolism (up to 40% VTE risk)
- Loss of anticoagulant proteins in urine: antithrombin III, proteins C and S
- Increased hepatic synthesis of procoagulant proteins (fibrinogen, factors V, VIII)
- Hyperlipidemia causes hyperviscosity; fibrinolytic inhibitors increase clot risk
- Most common sites: renal vein thrombosis, DVT, pulmonary embolism
- Arterial thrombosis also possible
2. Infection
- Loss of IgG and complement factors in urine
- Reduced T-cell function
- Increased risk of cellulitis, spontaneous bacterial peritonitis, pneumococcal infections
- Steroid therapy adds further immunosuppression
3. Atherosclerosis / Cardiovascular Disease
- Prolonged hyperlipidemia accelerates vascular disease
4. Protein malnutrition
- Loss of vitamin D-binding protein → vitamin D deficiency, secondary hyperparathyroidism
- Loss of transferrin → iron-deficiency anemia
- Loss of hormone-binding proteins → thyroid/hormonal disturbances
Diagnosis
- Urinalysis with microscopy: proteinuria, lipiduria, fatty casts, oval fat bodies (no RBC casts - those suggest nephritic)
- 24-hour urine or spot urine protein:creatinine ratio to quantify proteinuria
- Serum: albumin, lipid panel, creatinine, BUN, electrolytes, CBC
- Complement (C3, C4, CH50) - low in MPGN, lupus nephritis
- Serologies: ANA, anti-dsDNA (SLE), anti-PLA2R (primary membranous), SPEP (amyloid), hepatitis B/C serology, HIV
- Renal biopsy - indicated in adults (most cases); children usually treated empirically for MCD first
Treatment
General / Supportive
| Goal | Intervention |
|---|---|
| Edema | Loop diuretics (furosemide); restrict dietary Na+ (<2 g/day); fluid restriction if severe hyponatremia |
| Proteinuria reduction | ACE inhibitors or ARBs - reduce intraglomerular pressure, reduce proteinuria, slow CKD progression |
| Hyperlipidemia | Statins |
| Thromboembolism prophylaxis | Anticoagulation considered if serum albumin <2.5 g/dL or other high-risk features; long-term if renal vein thrombosis occurs |
| Infections | Pneumococcal and influenza vaccination; prophylactic antibiotics in selected cases |
Note on diuretic resistance: Hypoalbuminemia reduces protein-binding of furosemide, enlarging its volume of distribution and limiting delivery to tubular sites. ACE inhibitor/ARB addition reduces proteinuria and synergizes with loop diuretics.
Disease-Specific Treatment
| Cause | First-Line | Notes |
|---|---|---|
| Minimal Change Disease | Oral corticosteroids (prednisone 1 mg/kg/day, max 80 mg) | >80% achieve remission; relapses common; steroid-dependent cases use calcineurin inhibitors (cyclosporine, tacrolimus) or rituximab |
| FSGS | Corticosteroids | Often steroid-resistant; calcineurin inhibitors, mycophenolate, rituximab for resistant cases |
| Membranous Nephropathy | Observation for mild cases; immunosuppression for high-risk | Cyclophosphamide + steroids (Ponticelli regimen) or rituximab (now preferred for primary MN) |
| Diabetic Nephropathy | RAAS blockade; SGLT2 inhibitors | Address underlying glycemic and blood pressure control |
| Lupus Nephritis (Class V) | Mycophenolate mofetil ± steroids | Belimumab and voclosporin now adjunctive options |
KDIGO Pediatric Protocol (2021)
- Initial episode: Prednisone 60 mg/m²/day (max 60 mg/day) for 4-6 weeks, then 40 mg/m² on alternate days for another 4-6 weeks
- Relapse: Prednisone until remission, then taper
- Frequent relapsers/steroid-dependent: Calcineurin inhibitors (cyclosporine, tacrolimus) or mycophenolate mofetil; rituximab for refractory cases
Key Distinctions: Nephrotic vs. Nephritic
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Proteinuria | >3.5 g/day (massive) | Mild-moderate |
| Hematuria | Absent or mild | Present (RBC casts) |
| Hypertension | Variable | Prominent |
| Edema | Prominent | Mild |
| GFR | Normal or mildly reduced | Reduced (azotemia) |
| Complement | Usually normal | Often low (MPGN, post-strep, lupus) |
Recent Evidence Note
A 2025 Cochrane review (PMID 40337980) examined interventions for idiopathic steroid-resistant nephrotic syndrome in children - reflecting ongoing research into the management of the most challenging cases. The evidence base continues to evolve particularly for rituximab and calcineurin inhibitor use in steroid-dependent and steroid-resistant disease.
Sources: Comprehensive Clinical Nephrology 7th Ed.; Brenner & Rector's The Kidney; Robbins & Kumar Basic Pathology; Frameworks for Internal Medicine; Tintinalli's Emergency Medicine; Fishman's Pulmonary Diseases
What is the role of SGLT2 inhibitors in slowing nephrotic syndrome progression?
Searching PubMed
"SGLT2 inhibitor" AND "nephrotic syndrome"
Finding Sources
Searching PubMed
"SGLT2 inhibitor" AND proteinuria AND glomerular
Reading Article
PMID: 41203232
Reading Article
PMID: 41202026
Reading Article
PMID: 38061371
Reading File
Reading File
Searching PubMed
"SGLT2 inhibitor" AND "IgA nephropathy" OR "focal segmental glomerulosclerosis" OR "membranous nephropathy"
I now have excellent, current data from landmark trials and meta-analyses. Let me compile the comprehensive answer.
SGLT2 Inhibitors in Nephrotic Syndrome Progression
Background: What SGLT2 Inhibitors Do in the Kidney
SGLT2 (sodium-glucose cotransporter 2) inhibitors block glucose and sodium reabsorption in the S1 segment of the proximal tubule. In doing so, they produce several downstream effects relevant to proteinuric kidney disease:
Core Mechanisms of Renoprotection
| Mechanism | Detail |
|---|---|
| Tubuloglomerular feedback (TGF) restoration | By blocking proximal Na+ reabsorption, more Na+ and Cl- reach the macula densa → afferent arteriole constriction → reduced intraglomerular capillary pressure → reduced glomerular hyperfiltration |
| Reduced glomerular hyperfiltration | In diabetes, hyperfiltration damages glomeruli and drives proteinuria. SGLT2i reverses this; but the same TGF mechanism applies even in non-diabetic CKD |
| Reduction in proteinuria | Lower intraglomerular pressure directly reduces the hydraulic driving force for protein leakage through the glomerular filtration barrier |
| Anti-fibrotic / anti-inflammatory effects | Reduced tubular glucose burden lowers advanced glycation end products (AGEs) and pro-fibrotic cytokines (TGF-β, connective tissue growth factor) |
| Natriuresis and volume reduction | Reduces preload/afterload, lowers intraglomerular pressure via hemodynamic effects |
| Mitochondrial and metabolic effects | Promotes ketone body use, reduces oxidative stress in tubular cells; reduces mTOR signaling and tubular hypertrophy |
As Goldman-Cecil notes, SGLT2 inhibitors "reduce this elevated intraglomerular capillary pressure in experimental animals, so this hyperfiltration hypothesis potentially explains how these medications slow the progression of diabetic nephropathy" - and this mechanism is independent of glucose lowering.
Evidence Base: From Diabetic to Non-Diabetic Proteinuric Disease
Landmark Trials
| Trial | Drug | Population | Key Kidney Outcome |
|---|---|---|---|
| CREDENCE (2019) | Canagliflozin | T2DM + diabetic nephropathy (mean UACR ~927 mg/g) | 34% relative risk reduction in renal composite endpoint |
| DAPA-CKD (2020) | Dapagliflozin | CKD (eGFR 25-75) with UACR ≥200 mg/g; ~33% non-diabetic | 39% reduction in sustained eGFR decline, ESKD, or renal death; benefit preserved in non-diabetic patients |
| EMPA-KIDNEY (2022-2024) | Empagliflozin | Broad CKD including eGFR as low as 20; many non-diabetic | Halved the chronic eGFR decline rate from -2.75 to -1.37 mL/min/1.73m²/year (-50%, 95% CI 42-58%); largest relative benefit in those with lower baseline UACR |
DAPA-CKD: FSGS Pre-specified Subgroup
A pre-specified analysis of DAPA-CKD (PMID 34850160) specifically examined patients with focal segmental glomerulosclerosis - a primary cause of nephrotic syndrome. Dapagliflozin reduced the composite kidney endpoint with an HR consistent with the overall trial benefit, supporting efficacy in this non-diabetic proteinuric glomerular disease.
Cutting-Edge 2026 Meta-Analysis Evidence
Two landmark JAMA meta-analyses published in January 2026 (SMART-C Consortium, 70,000+ participants across 10 trials) provide the most current and robust picture:
1. Neuen et al., JAMA 2026 (PMID 41203232)
- SGLT2 inhibitors reduced CKD progression risk by 38% overall (HR 0.62, 95% CI 0.57-0.68) across 70,361 participants
- Benefit was preserved regardless of baseline eGFR - including stage 4 CKD (eGFR <30): HR 0.71
- Benefit was preserved regardless of albuminuria level - including those with >300 mg/g (nephrotic-range proteinuria): HR 0.57 (95% CI 0.52-0.64)
- Annual eGFR decline was slowed across all UACR subgroups, in both diabetic and non-diabetic patients
2. Staplin et al., JAMA 2026 (PMID 41202026)
- Benefits on kidney disease progression were clear in both diabetic and non-diabetic patients (HR 0.65 vs. 0.74)
- Absolute benefits were largest in those with UACR ≥200 mg/g - the heavy proteinuria range that overlaps with nephrotic syndrome
- Benefits also extended to reduced AKI risk, hospitalizations, and mortality
Specific Relevance to Nephrotic Syndrome Causes
Diabetic Nephropathy (most common secondary NS)
The strongest evidence base. SGLT2 inhibitors are now recommended alongside RAAS blockade as standard of care in diabetic nephropathy with proteinuria. Harrison's 22E (2025) states: "SGLT2 inhibitors in the proximal tubule of the kidney create glycosuria, improve glucose control, and have been shown to reduce the rate of progression" in diabetic nephropathy.
IgA Nephropathy
The 2023 case report (PMID 38318200) described efficacy in IgA nephropathy with nephrotic syndrome. Dapagliflozin is now specifically approved for IgA nephropathy based on DAPA-CKD data, and the June 2026 ALIGN trial (42242268) (atrasentan) suggests the field is moving toward combination approaches for this disease.
FSGS
The DAPA-CKD FSGS subgroup is the primary data source. Evidence supports using SGLT2 inhibitors as an adjunct to RAAS blockade to reduce proteinuria and slow eGFR decline, even though FSGS-specific immunotherapy remains the mainstay for active nephrotic syndrome.
Minimal Change Disease / Primary NS
Evidence is weakest here. MCD typically causes episodic nephrotic syndrome that responds to steroids. There is no RCT evidence specifically in MCD, and the transient nature of MCD episodes makes SGLT2i less relevant as a long-term renoprotective strategy compared to diseases like FSGS or membranous nephropathy. Current case-level evidence only.
Practical Role in the Nephrotic Syndrome Management Algorithm
Nephrotic Syndrome
|
├─ Identify underlying cause (biopsy, serologies)
|
├─ ALL patients with persistent proteinuria:
| ├─ RAAS blockade (ACE-i or ARB) ← first line
| └─ SGLT2 inhibitor ← now standard adjunct if eGFR ≥20
| (particularly if UACR still >200 mg/g on RAAS alone)
|
├─ Diabetic nephropathy: SGLT2i + RAAS + finerenone
├─ FSGS/IgA: SGLT2i + RAAS ± specific immunosuppression
└─ MCD: Steroids first; SGLT2i not established
Key Practical Points
- eGFR dip: An initial acute drop of ~2-3 mL/min/1.73m² is expected and acceptable - this reflects the TGF-mediated afferent arteriole constriction and does not indicate harm. The long-term chronic slope benefit is preserved.
- SGLT2i + RAAS combination is additive - each reduces intraglomerular pressure via different mechanisms
- Volume effects: SGLT2i-driven natriuresis can exacerbate volume depletion in underfill-type nephrotic syndrome (particularly MCD) - monitor closely
- Furosemide interaction: The natriuretic effect of SGLT2i may reduce diuretic requirements; dose adjustment may be needed
- Infections: Genital mycotic infections are an adverse effect; already immunocompromised nephrotic patients require vigilance
Summary
SGLT2 inhibitors have gone from a diabetes drug to a cornerstone of renoprotection in proteinuric CKD. The January 2026 SMART-C meta-analyses (70,000+ patients) confirm benefit across all eGFR and albuminuria strata, including in non-diabetic patients with heavy proteinuria. In nephrotic syndrome specifically:
- Strongest evidence: Diabetic nephropathy, IgA nephropathy, FSGS with CKD
- Mechanism: Primarily hemodynamic (TGF-mediated reduction in glomerular hyperfiltration) + anti-fibrotic + metabolic
- Not yet established: Primary MCD, MPGN, membranous nephropathy as dedicated indications
- Current guideline position: KDIGO 2024 recommends SGLT2 inhibitors for CKD with eGFR ≥20 and UACR ≥200 mg/g, regardless of diabetes status - which captures the majority of patients with clinically significant nephrotic syndrome
Sources: Harrison's Principles of Internal Medicine 22E (2025); Goldman-Cecil Medicine; Neuen et al. JAMA 2026 (PMID 41203232); Staplin et al. JAMA 2026 (PMID 41202026); EMPA-KIDNEY trial (PMID 38061371); DAPA-CKD FSGS subgroup (PMID 34850160)
sickle cell disease
Reading File
Finding Sources
Searching PubMed
"sickle cell disease"[MeSH Terms] AND management
Finding Sources
Searching PubMed
"sickle cell"[MeSH Terms] AND treatment
Finding Sources
Reading File
Reading File
Reading File
Searching PubMed
"sickle cell disease" AND (hydroxyurea OR voxelotor OR crizanlizumab OR gene therapy)
Reading File
Reading Article
PMID: 41831848
Loading Image
Loading Image
Reading File
Finding Sources
Reading Article
PMID: 39118728
Reading File
I have all the material I need for a comprehensive answer. Let me now compile it.
Sickle Cell Disease
Definition and Genetics
Sickle cell disease (SCD) is a group of inherited hemoglobinopathies caused by a single nucleotide mutation in the beta-globin gene (codon 6: adenine-to-thymine transversion, A6T) that substitutes valine for glutamic acid at position 6 of the beta-globin chain. This produces sickle hemoglobin (HbS) in place of normal HbA.
- Globally affects millions, particularly in sub-Saharan Africa, the Middle East, India, and the Mediterranean
- In the United States, ~8% of people of African descent are HbS carriers; ~1 in 600 have sickle cell anemia (HbSS)
- The HbS allele is maintained by selective pressure because heterozygous carriers are protected against falciparum malaria
Genotypes and Severity
| Genotype | Description | HbS% | Severity |
|---|---|---|---|
| HbSS (sickle cell anemia) | Homozygous; no HbA | >85% | Most severe |
| HbSS + α-thalassemia | Milder due to reduced MCHC | >85% | Moderate-severe |
| HbSC disease | HbS + HbC (Lys at β6) | 50% | Moderate |
| HbS β⁰-thalassemia | HbS + no β-globin production | >85% | Severe (= HbSS) |
| HbS β⁺-thalassemia | HbS + reduced β-globin | 70-95% | Mild-moderate |
| Sickle cell trait (HbAS) | Heterozygous carrier | 35-40% | Asymptomatic (usually) |
HbF (fetal hemoglobin, α₂γ₂) inhibits HbS polymerization - this is why newborns don't manifest disease until ~5-6 months of age when HbF falls to adult levels.
Pathophysiology
Step 1: HbS Polymerization
On deoxygenation, the valine residue at β6 creates a hydrophobic contact site allowing HbS molecules to self-assemble into long polymers. These polymers distort the red cell into the characteristic elongated crescentic (sickle) shape.
- Sickling is initially reversible on reoxygenation
- Repeated cycles of sickling cause cumulative membrane damage (Ca²+ influx, K+ and water loss) → irreversibly sickled cells prone to hemolysis
Factors that promote HbS polymerization:
- High intracellular HbS concentration
- Low pH (acidosis)
- High temperature
- Slow transit through the microvasculature (delayed deoxygenation)
Factors that inhibit polymerization:
- Presence of HbF or HbA (dilutes HbS, weak interaction)
- Good hydration (reduces intracellular HbS concentration)
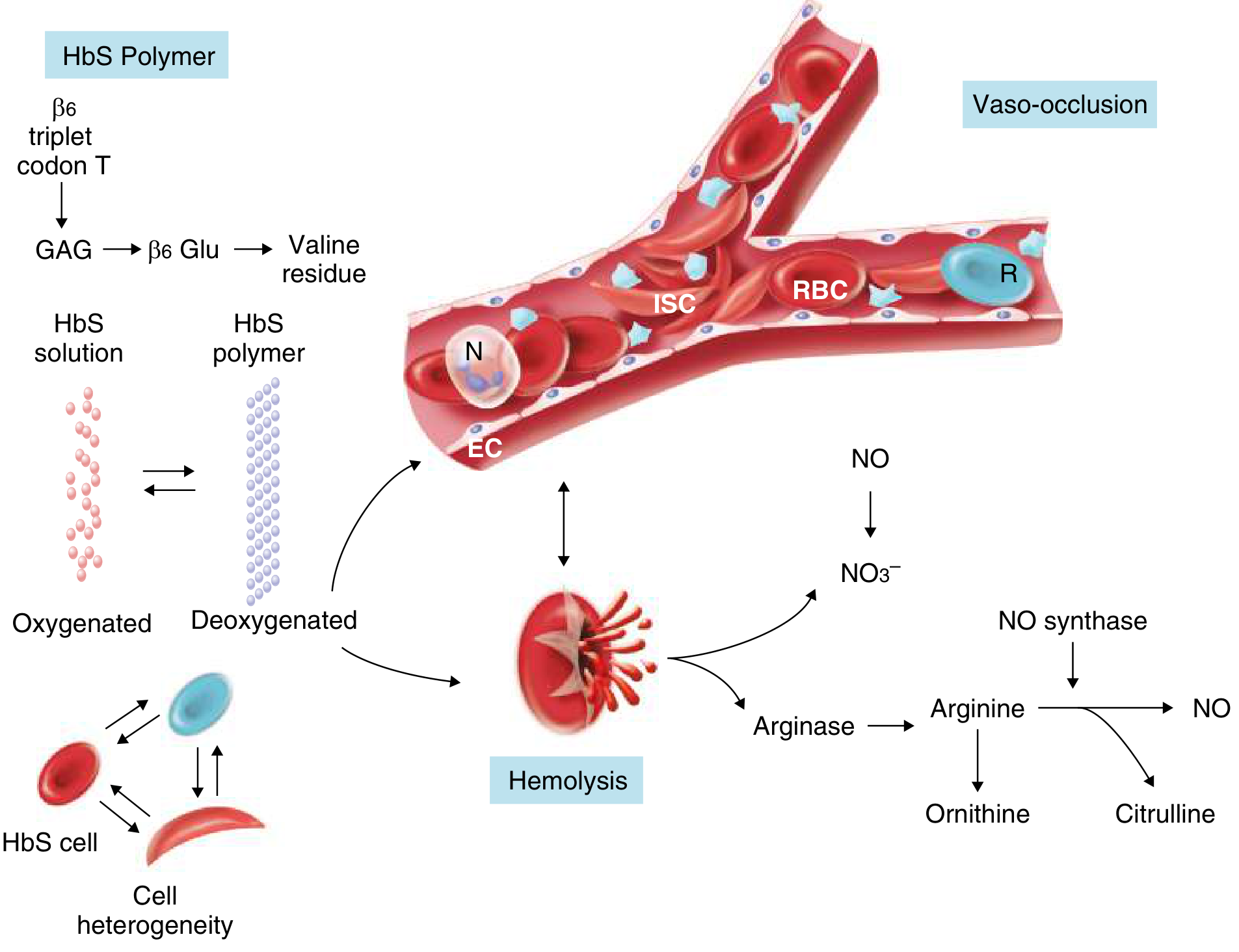
Step 2: Two Downstream Cascades
A. Hemolysis
- Extravascular: Macrophages phagocytose deformed sickled cells in the spleen and liver
- Intravascular: Direct red cell lysis releases free hemoglobin into plasma, which:
- Depletes haptoglobin
- Scavenges nitric oxide (NO) → vasoconstriction, platelet activation, endothelial inflammation
- Releases arginase → destroys L-arginine (the NO synthase substrate) → further reduces NO
- Produces reactive oxygen species
B. Vaso-Occlusion
Sickled cells adhere to activated vascular endothelium via cell adhesion molecules (P-selectin, VCAM-1), trapping leukocytes, platelets, and reticulocytes. This creates a cascade of:
- Microvascular obstruction → tissue ischemia
- Reperfusion injury → further endothelial activation
- Inflammation → more sickling
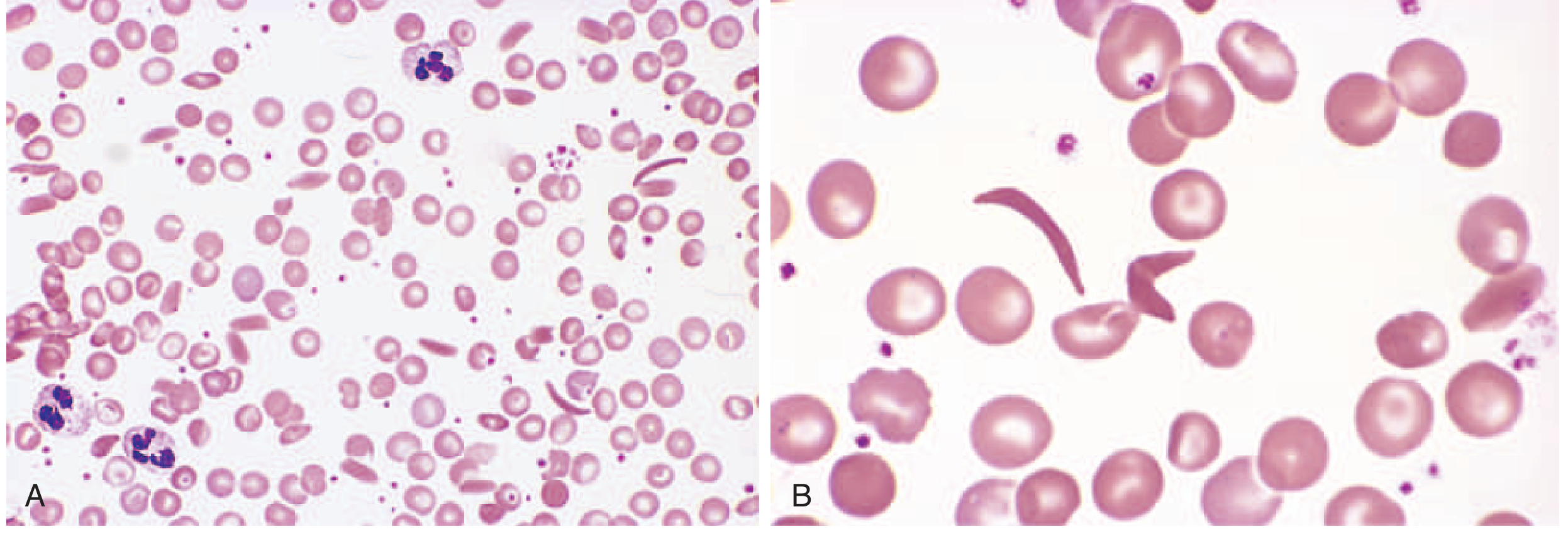
Clinical Manifestations
Acute Complications
1. Vaso-Occlusive Crisis (VOC) / Painful Crisis
- Most frequent acute complication; caused by microvascular occlusion
- Severe pain in bones (especially long bones, back, chest, abdomen)
- Triggers: cold, infection, dehydration, hypoxia, stress, alcohol
- Management: IV fluids, analgesia (NSAIDs + opioids), warmth, oxygen if hypoxic
2. Acute Chest Syndrome (ACS)
- New pulmonary infiltrate + fever + respiratory symptoms
- Caused by fat embolism (from bone marrow infarction), infection, or in-situ sickling in pulmonary vasculature
- Most common cause of death in SCD
- Treatment: broad-spectrum antibiotics + exchange transfusion + respiratory support; hydroxyurea for recurrent ACS
3. Stroke and Cerebrovascular Disease
- Children: ischemic stroke from large vessel intracranial stenosis
- Adults: silent infarcts, hemorrhagic stroke
- Transcranial Doppler (TCD) screening identifies high-risk children; chronic transfusion program reduces stroke risk by ~90%
- Hydroxyurea is an alternative to transfusion in lower-resource settings
4. Aplastic Crisis
- Triggered by Parvovirus B19 infection (infects erythroid precursors)
- Sudden severe drop in hemoglobin + reticulocytopenia
- Self-limited; supportive transfusion
5. Splenic Sequestration
- Rapid pooling of blood in the spleen → acute splenomegaly + severe anemia + cardiovascular collapse
- Most common in children (before functional asplenia develops)
- Emergency splenectomy or transfusion; elective splenectomy after recurrence
6. Acute Kidney Injury
- Occurs in 2-10% of acute painful crises
- Precipitated by dehydration, sepsis, drugs (NSAIDs)
Chronic Complications (Organ Damage)
| System | Complication |
|---|---|
| Spleen | Functional asplenia by age 5-6 years from repeated infarcts (autosplenectomy) → increased risk of encapsulated bacteria (pneumococcus, meningococcus, H. influenzae) |
| Bones | Avascular necrosis (AVN) of femoral/humeral heads; vertebral body H-shaped infarcts ("Lincoln log" sign); osteomyelitis (Salmonella classically) |
| Kidney | Hyposthenuria (inability to concentrate urine), microalbuminuria (20% of children, 60% of adults), proteinuria, nephrotic syndrome, renal papillary necrosis, renal medullary carcinoma, end-stage renal disease |
| Lungs | Pulmonary hypertension (30% of adults); restrictive lung disease (70% of adults); sleep-disordered breathing (40-60%) |
| Heart | High-output cardiac failure; dilated cardiomyopathy from chronic anemia |
| Eyes | Proliferative retinopathy (especially HbSC); vitreous hemorrhage; retinal detachment |
| CNS | Silent cerebral infarcts (35% of children by age 14); cognitive impairment; headaches |
| Liver | Sickle hepatopathy; intrahepatic cholestasis; pigment gallstones (from chronic hemolysis) in 50-70% |
| Skin | Leg ulcers (chronic, hard to heal; from local ischemia) |
| Genitourinary | Priapism (stuttering or fulminant) → risk of erectile dysfunction; enuresis |
| Immune | Increased COVID-19 severity; sepsis from encapsulated organisms |
Diagnosis
Blood Tests
- CBC: Hemoglobin 5-10 g/dL (HbSS); normochromic normocytic anemia; reticulocytosis (10-25%); leukocytosis; thrombocytosis
- Peripheral smear: Sickle cells, target cells, Howell-Jolly bodies (asplenia), nucleated RBCs
- Hemoglobin electrophoresis / HPLC: Gold standard for diagnosis; identifies HbS, HbA, HbF, HbA2 percentages
- Newborn screening: Now mandatory in most high-income countries (isoelectric focusing or HPLC of cord blood)
Additional Tests
- Elevated bilirubin, LDH (hemolysis markers)
- Low haptoglobin
- TCD ultrasound for stroke risk stratification
- Echocardiography (pulmonary hypertension screening via tricuspid regurgitation velocity)
Treatment
Disease-Modifying Therapy
1. Hydroxyurea (Hydroxycarbamide) - First-line
- Mechanism: Increases HbF production (inhibits ribonucleotide reductase, activates HbF gene transcription via stress erythropoiesis); reduces WBC and platelet adhesion; decreases MCHC
- Benefits: 50% reduction in painful crises, ACS, hospitalizations; improved survival; reduces renal hyperfiltration in children
- Dose: 15-35 mg/kg/day in adults; uptitrate to maximum tolerated dose
- Monitoring: CBC every 4-8 weeks (myelosuppression)
- Pregnancy: Teratogenic - contraindicated in pregnancy; use with effective contraception
- Administered under supervision of an experienced provider; now being expanded in low-resource settings
2. L-Glutamine
- FDA approved 2017; reduces oxidative stress in sickle cells; reduces VOC frequency
3. Crizanlizumab (Adakveo)
- Mechanism: Anti-P-selectin monoclonal antibody; blocks sickle cell-endothelium adhesion
- FDA approved 2019; reduces VOC frequency
- Note: In 2023, the FDA withdrew crizanlizumab's accelerated approval pending confirmatory trial data - its status as of 2026 requires current guidelines review
4. Voxelotor (Oxbryta)
- Mechanism: Allosterically stabilizes HbS in the oxygenated state, preventing polymerization; raises hemoglobin
- FDA approved 2019; also withdrawn from market in 2024 by manufacturer following safety data review - prescribers should check current status
5. Chronic Transfusion Program
- Monthly exchange or simple transfusions to maintain HbS <30%
- Indications: stroke prevention (TCD-positive children), recurrent ACS, recurrent VOC on hydroxyurea
- Risks: alloimmunization, iron overload (requires chelation), transfusion reactions
Curative Therapies
6. Allogeneic Hematopoietic Stem Cell Transplantation (HSCT)
- Only established cure prior to gene therapies
- Best results with HLA-matched sibling donor (<15% graft failure, >90% event-free survival in children)
- Expanding use of haploidentical and unrelated donors
- Risks: graft-versus-host disease, graft failure, transplant-related mortality
7. Gene Therapies (2023-2024 FDA Approvals)
Two gene therapies now FDA-approved for SCD in patients aged ≥12 years:
| Therapy | Mechanism | Brand |
|---|---|---|
| Exagamglogene autotemcel (exa-cel) | CRISPR/Cas9 editing - reactivates BCL11A enhancer → increases HbF | Casgevy (first-ever CRISPR therapy) |
| Betibeglogene autotemcel (beti-cel) | Lentiviral vector adds functional β-globin gene (βA-T87Q) | Zynteglo |
Both require myeloablative conditioning. Clinical trials showed near-elimination of VOC and ACS in treated patients. The 2026 Lancet review (Colombatti et al., PMID 41831848) confirms gene therapies are now approved for clinical use, with HSCT outcomes also improving through alternative donor strategies.
Supportive and Preventive Care
| Intervention | Details |
|---|---|
| Penicillin prophylaxis | From 2 months to at least 5 years (prevents pneumococcal sepsis in asplenic patients) |
| Vaccinations | Pneumococcal (PCV13 + PPSV23), meningococcal, H. influenzae, influenza, COVID-19 |
| Folic acid supplementation | 5 mg/day - supports increased erythropoiesis |
| Hydration | Prevents sickling by reducing intracellular HbS concentration |
| Pain management | NSAIDs + opioids (IV morphine for severe crises); avoid ketorolac >5 days; NSAIDS avoid after 30 weeks in pregnancy |
| Oxygen | Supplement only if hypoxic; routine O₂ does not prevent sickling in normoxic patients |
| Iron chelation | For transfusion-related iron overload (deferasirox orally; deferoxamine SC infusion) |
| TCD screening | Annual from age 2-16 years; chronic transfusion if velocity >200 cm/s |
| Ophthalmic screening | Annual from age 10 for retinopathy (especially HbSC) |
Special Situations
Pregnancy:
- Increased maternal mortality (6-fold vs. controls), preterm labor, placental infarction, SGA infants
- Hydroxyurea is contraindicated (teratogenic); NSAIDs avoided after 30 weeks
- Regional anesthesia preferred over general for cesarean
- Therapeutic transfusion goal: HbS <40%, Hb ~10 g/dL
Surgery:
- Preoperative transfusion to Hb 10 g/dL (simple transfusion, not exchange) reduces perioperative complications
- Maintain hydration, normothermia, normoxia
Prognosis
- Median life expectancy currently in the fifth decade for both sexes in high-income countries
- Major causes of death: ACS, infection, stroke, multiorgan failure
- Quality and length of life have improved significantly with hydroxyurea, transfusion programs, and now gene therapies
- The 2026 Lancet review identifies the transition from pediatric to adult care and equitable access to gene therapies as key ongoing challenges
Sources: Goldman-Cecil Medicine International Edition; Robbins & Kumar Basic Pathology; Harrison's Principles of Internal Medicine 22E (2025); Rosen's Emergency Medicine; Comprehensive Clinical Nephrology 7th Ed.; Colombatti et al. Lancet 2026 (PMID 41831848); Singh et al. Ann Med Surg 2024 (PMID 39118728)
REFEEDING SYNDROME
Finding Sources
Searching PubMed
"refeeding syndrome"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading Article
PMID: 40090863
Reading File
Loading Image
Reading Article
PMID: 39187889
Finding Sources
I have comprehensive content from multiple authoritative textbooks and current guidelines. I have the pathogenesis diagram. Now I have everything needed to write the full answer.
Refeeding Syndrome
Definition
Refeeding syndrome is a potentially life-threatening metabolic complication that occurs when nutrition (oral, enteral, or parenteral) is introduced or rapidly increased in a person who has been severely malnourished or starved for a prolonged period. It is defined by the emergence of electrolyte imbalances - principally hypophosphatemia, alongside hypokalemia and hypomagnesemia - accompanied by fluid shifts and thiamine deficiency, triggered by the resumption of carbohydrate intake and the resulting surge in insulin secretion.
First described after World War II, when severely malnourished Japanese prisoners of war were refed and suffered sudden electrolyte abnormalities and unexpected deaths.
Pathophysiology
During Starvation
In prolonged starvation, the body shifts from carbohydrate metabolism to using fatty acids and amino acids as primary fuel (gluconeogenesis, lipolysis). During this catabolic state:
- Insulin secretion is low
- Intracellular electrolytes (phosphate, potassium, magnesium) become severely depleted from cells due to lack of intake and ongoing losses
- Serum levels of phosphate, potassium, and magnesium can appear normal because bone mineral stores release phosphate to maintain a near-normal extracellular concentration - this masks the true total body depletion
- Thiamine (vitamin B1) stores are also depleted
On Reintroduction of Carbohydrates
When carbohydrates are introduced, glucose drives a rapid and large surge in insulin secretion, which activates multiple cellular transport mechanisms simultaneously:
- Na+/K+ ATPase is stimulated → potassium (and sodium) are driven intracellularly → hypokalemia
- Phosphate is consumed for glycolysis (synthesis of ATP, glucose-6-phosphate, fructose-1,6-bisphosphate) and rapidly shifts intracellularly → hypophosphatemia (the hallmark)
- Magnesium shifts intracellularly via incompletely understood mechanisms; hypomagnesemia also exacerbates hypokalemia (Mg²+ is required for K+ transport)
- Insulin promotes Na+ and water retention → volume expansion → dilutional electrolyte drop + risk of pulmonary edema and heart failure
- Thiamine is consumed as a cofactor for pyruvate dehydrogenase in the reactivated Krebs cycle → if already depleted, pyruvate cannot enter the cycle → Wernicke encephalopathy
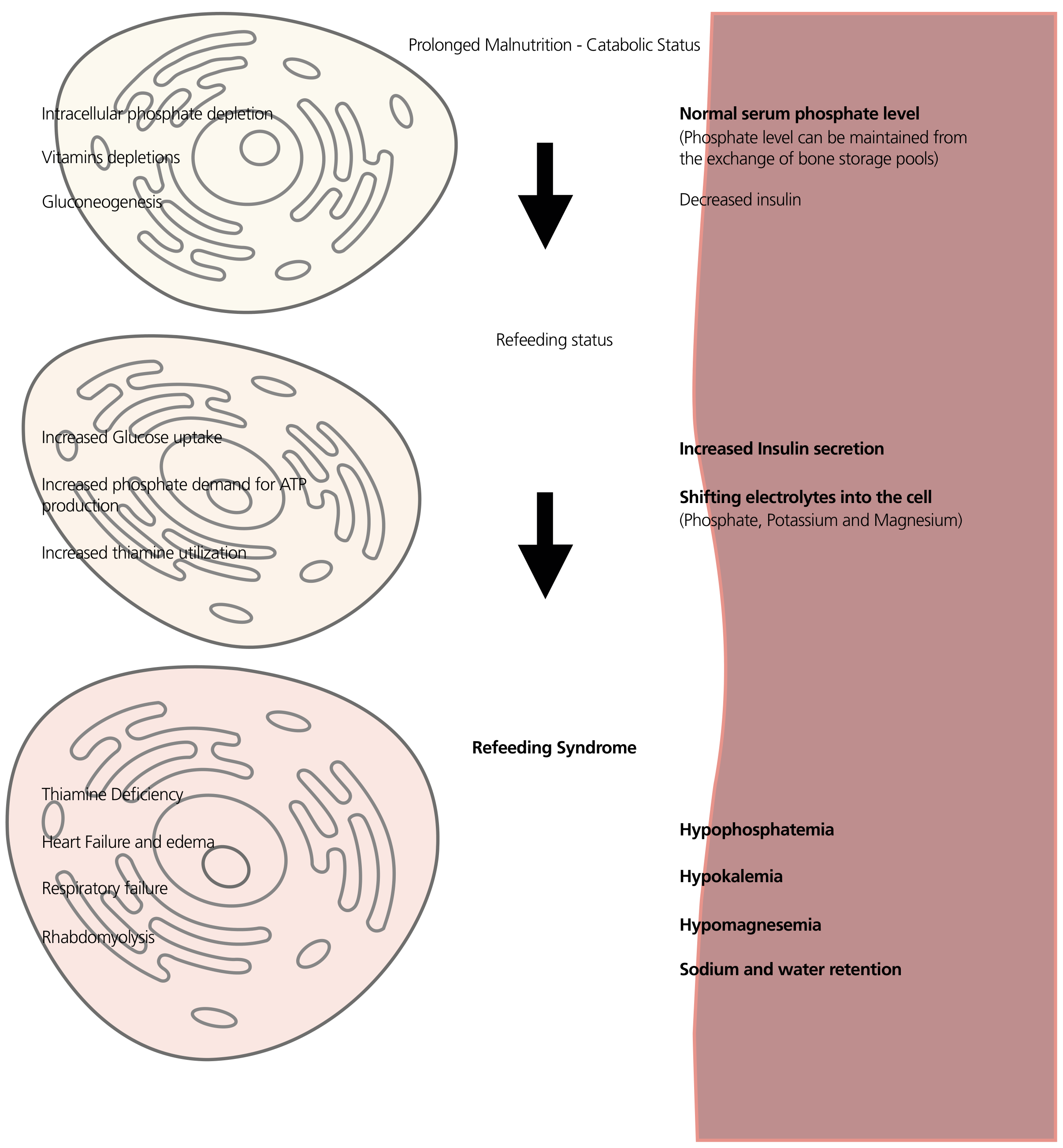
The central electrolyte: Phosphate is the linchpin because phosphorylated intermediates are needed for virtually every energy-producing reaction. When serum phosphate crashes, the body cannot produce ATP adequately, leading to dysfunction in every organ system.
Who is at Risk? (NICE Criteria)
High Risk - One criterion is sufficient:
| Criterion | Threshold |
|---|---|
| BMI | <16 kg/m² |
| Unintentional weight loss | >15% in the last 3-6 months |
| Nutritional intake | Little or none for >10 days |
| Pre-feeding electrolytes | Low potassium, phosphate, or magnesium before feeding |
Moderate Risk - Two or more criteria needed:
| Criterion | Threshold |
|---|---|
| BMI | <18.5 kg/m² |
| Unintentional weight loss | >10% in 3-6 months |
| Nutritional intake | Little or none for >5 days |
| Medications/substances | Insulin, chemotherapy, antacids, diuretics, alcohol misuse |
Clinical Conditions Commonly Associated with High Risk
- Anorexia nervosa (prolonged severe restriction)
- Chronic alcoholism (poor intake + thiamine depletion)
- Cancer (cachexia, chemotherapy, poor intake)
- Post-bariatric surgery (malabsorption, restriction)
- Prolonged nil-by-mouth (ICU, post-surgical, dysphagia)
- Chronic malnutrition from inflammatory bowel disease, short bowel syndrome, malabsorption
- Elderly patients with poor intake or acute illness
- HIV/AIDS
A 2025 systematic review (Zheng et al., PMID 39187889) identified the following as independently associated risk factors across 30 studies: alcohol misuse history, cancer, hypertension, high APACHE II or SOFA scores, low GCS, diuretic use before refeeding, low baseline prealbumin, elevated baseline creatinine, and enteral nutrition use.
Clinical Features
Symptoms typically emerge within 24-72 hours of initiating or increasing nutritional support.
Electrolyte Disturbances and Their Effects
| Electrolyte | Consequence |
|---|---|
| Hypophosphatemia (hallmark) | Muscle weakness, respiratory muscle failure (diaphragm), cardiac arrhythmias, heart failure, hemolysis (depletion of 2,3-DPG), rhabdomyolysis, seizures, coma |
| Hypokalemia | Cardiac arrhythmias (VT/VF), muscle weakness, ileus, respiratory failure |
| Hypomagnesemia | Arrhythmias, tetany, seizures, exacerbates hypokalemia and hypocalcemia |
| Hypocalcemia | Tetany, seizures, prolonged QT |
Other Features
- Sodium and water retention (insulin effect) → peripheral edema, acute heart failure, pulmonary edema
- Hyperglycemia → osmotic diuresis, worsening electrolyte losses
- Thiamine (B1) deficiency → Wernicke encephalopathy (ophthalmoplegia, ataxia, confusion) or Wernicke-Korsakoff syndrome; also cardiovascular beriberi (wet beriberi - high-output cardiac failure)
- Rhabdomyolysis → acute kidney injury
- Respiratory failure → ventilator dependence in ICU patients
At its most severe, the syndrome is fatal - cardiac arrest from ventricular arrhythmia or respiratory arrest.
Diagnosis
No universally accepted diagnostic criteria exist. The 2025 AuSPEN Consensus (Matthews-Rensch et al., PMID 40090863) states that refeeding syndrome should only be diagnosed if:
- The patient has had adequate nutrition intake (≥50% of estimated requirements) commenced
- Electrolyte imbalances emerge after its commencement
- Clinical symptoms are present
Laboratory Monitoring
Monitor the following before and daily for the first week after initiating feeding in at-risk patients:
| Electrolyte | Threshold of concern |
|---|---|
| Serum phosphate | <0.6 mmol/L = severe hypophosphatemia |
| Serum potassium | <3.5 mmol/L |
| Serum magnesium | <0.7 mmol/L |
| Serum calcium | Low |
| Blood glucose | Hyperglycemia / hypoglycemia |
| Thiamine | Clinical assessment; supplement preemptively |
| ECG | Arrhythmia detection |
| Fluid balance | Daily weight, intake/output |
Prevention and Management
Step 1 - Correct Pre-Existing Deficiencies BEFORE Feeding
- Replace low phosphate, potassium, and magnesium to normal levels before starting nutrition
- Give thiamine 100-200 mg IV/IM before initiating feeds or any glucose-containing fluids - this is non-negotiable
- Continue thiamine 100 mg/day for 5-7 days (longer in alcoholics and severely starved patients)
- Give multivitamins and trace elements
Step 2 - Start Nutrition Slowly
The cornerstone of prevention is a gradual, controlled reintroduction of calories:
| Risk Level | Starting Rate |
|---|---|
| Standard at-risk patients | Start at ~50% of estimated requirements on day 1 |
| Highest risk (prolonged starvation, chronic electrolyte losses) | Start at 10 kcal/kg/day maximum |
| Escalation | Increase gradually to meet full needs over 4-7 days |
Important caveat from AuSPEN 2025: There is no evidence that at-risk patients need to start at a lower enteral feeding rate than already recommended for checking GI tolerance. Goal nutrition rates should be reached within 24-72 hours for all routes. The old dogma of prolonged slow titration is being revised - but the 10 kcal/kg/day starting point remains standard for the highest-risk patients.
For parenteral nutrition (PN): start at 50% of the dextrose content on day 1 in at-risk patients.
Step 3 - Monitor Electrolytes Aggressively and Replace
- Check electrolytes (phosphate, potassium, magnesium, glucose) daily during the first week
- Replace electrolytes aggressively according to severity:
| Deficiency | Replacement Approach |
|---|---|
| Phosphate | Mild-moderate: oral sodium/potassium phosphate; Severe (<0.32 mmol/L): IV phosphate infusion. Select potassium-phosphate vs. sodium-phosphate based on concurrent potassium level |
| Potassium | Oral or IV; IV via central line for severe hypokalemia |
| Magnesium | Oral magnesium for mild; IV for severe; repleting Mg also helps correct refractory hypokalemia |
If electrolytes fall significantly during feeding, pause or reduce the rate and replace before recommitting.
Step 4 - Multidisciplinary Approach
The AuSPEN 2025 consensus emphasizes that "identification and management of refeeding syndrome requires a multidisciplinary approach" including:
- Nutrition support team (dietitian, gastroenterologist/intensivist)
- Pharmacist (electrolyte replacement protocols)
- Nursing (monitoring, fluid balance)
- Medicine/ICU (cardiac monitoring, respiratory support if needed)
Specific Routes of Nutrition
| Route | Key Points |
|---|---|
| Enteral nutrition (EN) | Risk is the same as PN; does not confer protection. Enteral nutrition was independently associated with higher RFS risk in the Zheng 2025 systematic review - likely due to more patients receiving EN overall |
| Parenteral nutrition (PN) | Higher incidence historically (up to 1 in 3 in some series); start at 50% dextrose on day 1 |
| Oral refeeding | Still carries risk; gradual reintroduction with supplementation required |
Special Populations
ICU patients: High risk due to prior nutritional deficits, organ dysfunction, and use of diuretics. Hypophosphatemia in critically ill patients correlates with increased mortality, ventilator duration, and SOFA scores. A 2026 systematic review of risk prediction models in critically ill adults (Dai et al., PMID 41793849) found high APACHE II and SOFA scores as major predictors.
Post-bariatric surgery: A 2025 systematic review (Triantafyllidis et al., PMID 40900247) specifically identified this as an underrecognized high-risk scenario.
Children: The Harriet Lane Handbook notes refeeding syndrome in pediatric patients - particularly important in eating disorders, oncology, and post-surgical malnutrition. Supplementary thiamine before feeds is recommended.
Anorexia nervosa: Among the highest-risk groups. Serum phosphate drops are expected and can be profound. Inpatient refeeding under close monitoring is preferred.
Chronic alcoholism: Concurrent thiamine depletion makes Wernicke encephalopathy a major risk. IV thiamine must be given before glucose - glucose infusion in a thiamine-depleted patient can precipitate acute Wernicke's.
Summary: The "Refeeding Checklist"
Before feeding a malnourished patient:
1. Assess risk (NICE criteria)
2. Check baseline electrolytes (PO4, K+, Mg2+, Ca2+)
3. Correct deficiencies BEFORE starting
4. Give thiamine 100-200mg IV before any glucose/feeds
5. Start at 10 kcal/kg/day if highest risk; ~50% of needs if moderate risk
6. Monitor daily electrolytes + glucose for first week
7. Replace electrolytes aggressively as needed
8. Escalate calories gradually over 4-7 days
9. Ensure multidisciplinary oversight
Sources: Bailey & Love's Short Practice of Surgery 28th Ed.; Yamada's Textbook of Gastroenterology 7th Ed.; Sabiston Textbook of Surgery; Tietz Textbook of Laboratory Medicine 7th Ed.; Matthews-Rensch et al. AuSPEN Consensus 2025 (PMID 40090863); Zheng et al. Nutr Clin Pract 2025 (PMID 39187889); Triantafyllidis et al. Obes Surg 2025 (PMID 40900247)
This is a shared conversation. Sign in to Orris to start your own chat.