1. Biosynthesis of catecholamines steps & enzymes 2. Mechanism of histamine action + provide cases of food poisoning 3. Which compounds inactivate peptide neurotransmitters 4. Proteins and neurotransmitters in Alzheimer’s disease 5. Catecholamines transporters 5.1 Which drug increase this transporter. 6. Function and characteristics of excitatory neurotransmitters 7. Function and properties of endocannabinoids 8. Mechanism and symptoms of Parkinson’s disease. These are 8 biochemistry open questions can you give me answers according to biochemistry in detail for second semester final exam
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Here is your comprehensive biochemistry exam guide covering all 8 questions, sourced directly from Harper's, Kaplan & Sadock, Lippincott, Goodman & Gilman, Guyton & Hall, and Stahl's Psychopharmacology.
Biochemistry - 8 Open Questions: Full Exam Guide
1. Biosynthesis of Catecholamines: Steps & Enzymes
Dopamine, norepinephrine, and epinephrine are all synthesized from the amino acid L-tyrosine through a shared pathway. Tyrosine itself is derived from dietary intake or from phenylalanine via phenylalanine hydroxylase (a BH4-requiring enzyme) in the liver.
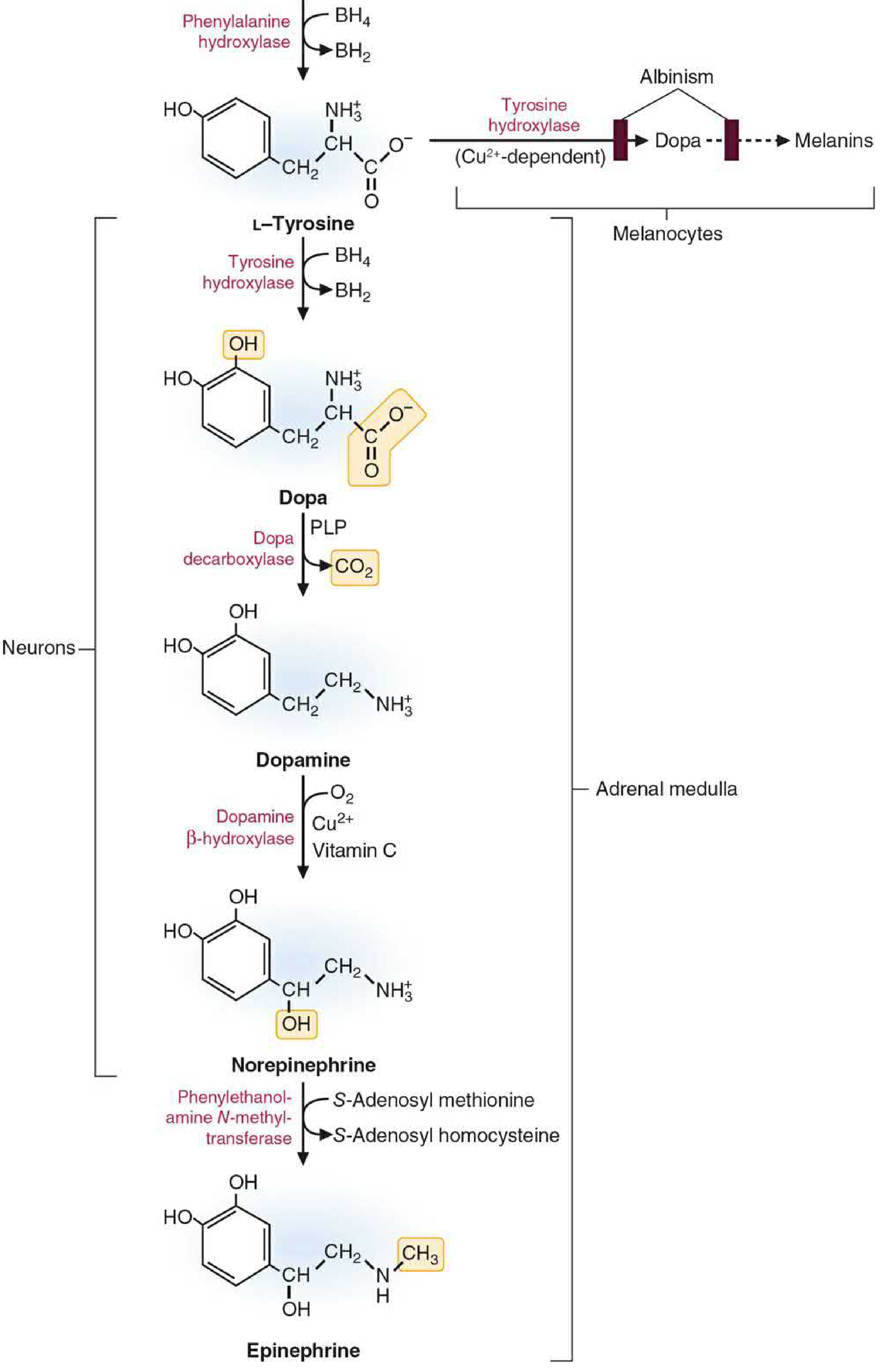
Step-by-Step Pathway
| Step | Substrate | Product | Enzyme | Cofactors |
|---|---|---|---|---|
| 1 (Rate-limiting) | L-Tyrosine | L-DOPA (dihydroxyphenylalanine) | Tyrosine hydroxylase (TH) - encoded by TH gene | Tetrahydrobiopterin (BH4), O2 |
| 2 | L-DOPA | Dopamine | DOPA decarboxylase (AADC) - aromatic amino acid decarboxylase | Pyridoxal phosphate (PLP/Vitamin B6) |
| 3 | Dopamine | Norepinephrine | Dopamine β-hydroxylase (DBH) - inside vesicles only | Ascorbic acid (Vitamin C), Cu2+, O2 |
| 4 | Norepinephrine | Epinephrine | PNMT - phenylethanolamine N-methyltransferase | SAM (S-adenosyl methionine); requires B12 + folate |
Key Points for Exam
- Tyrosine hydroxylase is the rate-limiting step - it is a mixed-function oxidase regulated by end-product (catecholamine) feedback inhibition. Catecholamines compete with BH4 for the enzyme's cofactor-binding site. Depolarization activates kinases (PKA, PKC, CaM kinases) that phosphorylate TH, making it less sensitive to feedback.
- Step 2 (AADC) uses PLP and is the same enzyme that decarboxylates 5-HTP to serotonin. Dopaminergic neurons stop at dopamine - they do not have DBH or PNMT.
- Step 3 (DBH) occurs exclusively inside storage vesicles (not in the cytosol), requires vitamin C (ascorbic acid) as electron donor, and Cu2+ as a bound cofactor. Disulfiram (Antabuse) inhibits DBH.
- Step 4 (PNMT) depends on SAM for methyl donation, making it dependent on adequate vitamin B12 and folate (both needed to regenerate SAM from homocysteine).
- L-DOPA is the principal drug for Parkinson's disease - it bypasses the rate-limiting TH step and is converted to dopamine by AADC in the brain.
Sources: - Basic Medical Biochemistry 6e, p. 1632-1635 | Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 418-420
2. Mechanism of Histamine Action + Food Poisoning Cases
Biosynthesis & Release
Histamine is synthesized from the amino acid histidine by the enzyme histidine decarboxylase (requires PLP). It is stored in vesicles via VMAT (vesicular monoamine transporter) and released by exocytosis from mast cells, basophils, and histaminergic neurons.
Four Receptor Types - Mechanisms & Effects
| Receptor | G-Protein / Signal | Main Location | Key Effects |
|---|---|---|---|
| H1 | Gq → PLC → IP3/DAG → ↑Ca2+ | Bronchial smooth muscle, blood vessels, gut, CNS neurons | Bronchoconstriction, itching, nasal secretion, ↑wakefulness/appetite inhibition, gut contraction. Blocked by antihistamines (diphenhydramine, loratadine) |
| H2 | Gs → adenylyl cyclase → ↑cAMP | Gastric parietal cells, CNS | Stimulates gastric acid secretion (primary driver). Also: feedback inhibition of histamine release from mast cells. Blocked by cimetidine, famotidine (PPU drugs) |
| H3 | Gi → ↓cAMP | CNS (basal ganglia, hippocampus, cortex) - presynaptic autoreceptors | Inhibits histamine release (autoreceptor). Also acts as heteroreceptor to modulate serotonin, dopamine, GABA, glutamate, NE. H3 agonists → sleep; H3 antagonists → wakefulness |
| H4 | Gi → ↓cAMP | Eosinophils, mast cells, T cells, GI tract | Immune and inflammatory responses; reduced expression in some cancers (breast, colorectal, gastric) |
Cardiovascular & Other Actions
- Vasodilation of resistance vessels (both H1 and H2) → lowers blood pressure; increases capillary permeability → edema formation
- Bronchoconstriction via H1 in lungs (key in anaphylaxis)
- Sensory nerves - stimulates itching via H1
Inactivation of Histamine
- In the brain: Histamine methyltransferase (HNMT) methylates histamine using SAM → N-methylhistamine → then oxidized by MAO-B
- In peripheral tissues: Diamine oxidase (DAO) deaminates histamine → then oxidized to a carboxylic acid
- Note: there is no reuptake transporter for histamine (unlike monoamines)
Food Poisoning Cases Involving Histamine
Scombroid fish poisoning (Histamine fish poisoning):
- Occurs when poorly refrigerated dark-flesh fish (tuna, mackerel, mahi-mahi, sardines) decompose. Bacteria (Morganella morganii, Klebsiella pneumoniae) decarboxylate histidine to histamine via bacterial histidine decarboxylase.
- Symptoms within minutes to 1-2 hours: flushing, rash/urticaria, nausea, vomiting, diarrhea, headache, palpitations, burning sensation in the mouth, bronchoconstriction.
- Mimics an allergic reaction but is a direct pharmacological effect of excessive histamine absorption.
- Treatment: antihistamines (H1 + H2 blockers).
Fermented food poisoning:
- Improperly fermented cheeses, wine, beer, and sauerkraut can accumulate high histamine levels.
- In susceptible individuals with low intestinal diamine oxidase (DAO) activity, dietary histamine is not degraded and enters systemic circulation.
- Symptoms: headache, flushing, GI distress, palpitations.
Sources: - Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 1512-1523 | Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 421-434
3. Compounds that Inactivate Peptide Neurotransmitters
Unlike classical small-molecule neurotransmitters (dopamine, GABA, glutamate), peptide neurotransmitters are NOT inactivated by reuptake transporters. Their inactivation is entirely enzymatic, carried out by extracellular peptidases/proteases located on the plasma membrane surface or in the extracellular space.
Main Classes of Inactivating Enzymes
| Enzyme Class | Examples | Mechanism | Target Peptides |
|---|---|---|---|
| Endopeptidases | Neprilysin (NEP / enkephalinase / CD10), Endothelin-converting enzymes | Cleave internal peptide bonds | Enkephalins, substance P, neuropeptide Y, VIP, ANP |
| Aminopeptidases | Aminopeptidase N (APN), Leucine aminopeptidase | Remove N-terminal amino acids sequentially | Enkephalins, neurotensin, oxytocin fragments |
| Angiotensin-converting enzyme (ACE) | ACE (kininase II) | Dipeptidyl carboxypeptidase - removes dipeptides from C-terminus | Angiotensin I → Angiotensin II; inactivates bradykinin |
| Dipeptidyl peptidase IV (DPP-IV) | DPP-IV (CD26) | Cleaves after proline at position 2 from N-terminus | Glucagon-like peptide-1 (GLP-1), neuropeptide Y, substance P |
| Prolyl oligopeptidase (POP) | Prolyl endopeptidase | Cleaves at Pro residues | Thyrotropin-releasing hormone (TRH), oxytocin, vasopressin |
| Carboxypeptidases | Carboxypeptidase E/H | Remove C-terminal amino acids | Involved in post-translational processing |
Key Characteristics of Peptide NT Inactivation
- No reuptake - unlike dopamine, serotonin, GABA (which have specific plasma membrane transporters), peptides have no known reuptake mechanism after release.
- Enzymatic cleavage is irreversible - the signal ends when the peptide is degraded.
- Enzymes are ectoenzymes - located on the outer surface of the plasma membrane (e.g., neprilysin on postsynaptic neurons, glia, and endothelial cells), enabling them to act at the synapse or at some distance (volume transmission).
- Diffusion also contributes - peptides diffuse away from the release site and are eventually cleared.
- Different proteases for different peptides - each neuropeptide's inactivation involves a specific set of enzymes depending on its structure.
Clinical Relevance
- Enkephalinase (neprilysin) inhibitors (e.g., sacubitril in combination with valsartan = Entresto) were originally developed to reduce enkephalin degradation for analgesia; sacubitril is now used in heart failure because neprilysin also degrades ANP/BNP.
- DPP-IV inhibitors (sitagliptin, saxagliptin) are used in type 2 diabetes because they prevent degradation of GLP-1.
- ACE inhibitors (captopril, enalapril) block angiotensin II formation and preserve bradykinin, used for hypertension and heart failure.
Sources: - Kaplan & Sadock's Comprehensive Textbook of Psychiatry (Neuropeptides chapter) | Basic Medical Biochemistry 6e
4. Proteins and Neurotransmitters in Alzheimer's Disease
Alzheimer's disease (AD) has three defining pathological features that biochemists must know:
A. Amyloid Beta (Aβ) Plaques
- Precursor protein: Amyloid Precursor Protein (APP) - a transmembrane glycoprotein encoded by a gene on chromosome 21.
- Normal processing: APP is cleaved by α-secretase (non-amyloidogenic pathway) → produces soluble sAPPα.
- Pathological processing: APP is first cleaved by β-secretase (BACE1), then by γ-secretase (a complex containing presenilin-1 or presenilin-2) → produces Aβ40 and Aβ42 peptides. Aβ42 is more hydrophobic and prone to aggregation.
- Aβ peptides aggregate into oligomers (most neurotoxic form), then fibrils, then amyloid plaques (neuritic/senile plaques).
- Plaques accumulate extracellularly, also depositing in cerebral blood vessel walls (cerebral amyloid angiopathy).
B. Neurofibrillary Tangles (NFTs) - Tau Protein
- Tau is a microtubule-associated protein that normally stabilizes microtubules in neurons.
- In AD, tau becomes hyperphosphorylated due to an imbalance between protein kinases (GSK-3β, CDK5) and phosphatases (PP2A).
- Hyperphosphorylated tau detaches from microtubules → microtubules disassemble → free tau aggregates into paired helical filaments (PHFs) → NFTs form intracellularly.
- This disrupts axonal transport, causes neuroinflammation, and contributes to neuronal death.
C. Neurotransmitter Deficits
| Neurotransmitter | Change | Pathway Affected | Clinical Consequence |
|---|---|---|---|
| Acetylcholine | Severely reduced | Nucleus basalis of Meynert → cortex and hippocampus | Memory loss, cognitive decline - the most prominent NT deficit. Correlates with degree of dementia |
| Glutamate | Dysregulated - excess synaptic glutamate | Cortical and hippocampal pathways | Excitotoxicity via overstimulation of NMDA receptors → Ca2+ influx → neuronal death |
| Serotonin | Reduced | Raphe nuclei projections | Depression, behavioral symptoms |
| Norepinephrine | Reduced | Locus coeruleus projections | Attention and cognitive deficits |
D. Other Key Proteins
- ApoE4 (apolipoprotein E4) - the major genetic risk factor for sporadic AD. ApoE4 impairs Aβ clearance and promotes tau phosphorylation. ApoE4 homozygosity increases risk ~12x vs. ApoE3.
- Presenilin-1 (PSEN1) and Presenilin-2 (PSEN2) - mutations cause familial early-onset AD by altering γ-secretase activity to overproduce Aβ42.
- Alpha-synuclein - primarily associated with Parkinson's but can co-occur in AD (Lewy body variant).
E. Current Drug Targets
- AChE inhibitors (donepezil, galantamine, rivastigmine): increase ACh by blocking acetylcholinesterase
- NMDA antagonist (memantine): blocks overactivated NMDA receptors to reduce glutamate excitotoxicity
- Anti-amyloid monoclonal antibodies (lecanemab, donanemab): remove Aβ plaques; slow but do not halt progression
Sources: - Lippincott Pharmacology 6e, p. 531 | Guyton & Hall Textbook of Medical Physiology, p. 751 | Kaplan & Sadock's, p. 443
5. Catecholamine Transporters
5.1 Plasma Membrane Transporters (SLC6 Gene Family) - for Reuptake
These transporters terminate neurotransmitter action by removing monoamines from the synapse back into the presynaptic neuron. They use secondary active transport - co-transport with Na+ ions (driven by the Na+/K+-ATPase gradient). Some also co-transport Cl- and counter-transport K+.
| Transporter | Full Name | Monoamine | Location |
|---|---|---|---|
| DAT | Dopamine Transporter | Dopamine (primary); also amphetamine | Dopaminergic neurons in striatum, nucleus accumbens, substantia nigra; high density in these areas |
| NET | Norepinephrine Transporter | Norepinephrine (primary); also dopamine (high affinity for DA too) | Noradrenergic neurons, locus coeruleus projections, prefrontal cortex |
| SERT | Serotonin Transporter | Serotonin | Serotonergic neurons (raphe nuclei) |
Note: NET has high affinity for dopamine, making it a major route of dopamine reuptake in the prefrontal cortex (where DAT density is low).
5.2 Vesicular Transporters (SLC18 Gene Family)
| Transporter | Full Name | Function |
|---|---|---|
| VMAT2 | Vesicular Monoamine Transporter 2 | Packages all three catecholamines AND serotonin AND histamine from cytosol into synaptic vesicles. Used by ALL monoamine neurons. Mechanism: secondary active transport driven by a proton gradient (V-ATPase pumps H+ into vesicle; H+ exchanges for the monoamine). Intravesicular concentration ~100x cytosolic. |
5.3 Drugs That Increase (Block) These Transporters
"Increase the transporter" in the pharmacological sense means inhibit reuptake (block the transporter), which increases monoamine levels in the synapse.
| Drug | Transporter Blocked | Mechanism & Use |
|---|---|---|
| Cocaine | DAT, NET, SERT (all three) | Blocks all monoamine reuptake; euphoric effects from nucleus accumbens DA surge; addictive |
| Amphetamine / Methamphetamine | DAT, NET | Blocks reuptake AND causes active release of DA (reverse transport); used for ADHD; addictive |
| Methylphenidate (Ritalin) | DAT, NET | Blocks reuptake; ADHD treatment |
| Lisdexamfetamine (Vyvanse) | DAT, NET | Prodrug; cleaved to D-amphetamine in blood; long-acting ADHD treatment |
| TCAs (tricyclic antidepressants) | NET (primary), SERT | Block NE and serotonin reuptake; antidepressants |
| SNRIs (duloxetine, venlafaxine) | NET + SERT | Dual NE/serotonin reuptake inhibitors; depression, anxiety |
| SSRIs (fluoxetine, sertraline) | SERT | Selective serotonin reuptake inhibitors; depression |
| Bupropion (NDRI) | NET + DAT (weak) | Norepinephrine-dopamine reuptake inhibitor; depression, smoking cessation |
| Reserpine | VMAT2 (blocks vesicular uptake) | Depletes all monoamine stores; was used for hypertension; causes depression as a side effect |
Sources: - Stahl's Essential Psychopharmacology, p. 47-50 | Kaplan & Sadock's Comprehensive Textbook, p. 156-175 | Basic Medical Biochemistry 6e, p. 1634
6. Function and Characteristics of Excitatory Neurotransmitters
Major Excitatory Neurotransmitters
A. Glutamate (Primary Excitatory NT of the CNS)
- Synthesis: From α-ketoglutarate (TCA cycle intermediate) via transamination by aspartate aminotransferase (AAT), or from glutamine by glutaminase. Glutamate synthesis directly "drains" α-KG from the TCA cycle and must be replenished via anaplerotic reactions (pyruvate carboxylase, methylmalonyl-CoA mutase).
- Storage: Vesicular glutamate transporters (vGluT1-3, SLC17 family) package glutamate into synaptic vesicles.
- Receptors:
| Receptor Type | Type | Mechanism | Function |
|---|---|---|---|
| AMPA | Ionotropic | Na+/K+ channel (fast) | Fast synaptic excitation; mediates most excitatory neurotransmission |
| NMDA | Ionotropic | Na+/K+/Ca2+ channel; requires both glutamate AND glycine co-agonist; blocked by Mg2+ at rest | Long-term potentiation (LTP), memory, learning; Ca2+ influx crucial for plasticity |
| Kainate | Ionotropic | Na+/K+ channel | Modulates synaptic transmission |
| mGluR (1-8) | Metabotropic (GPCR) | Various G proteins → cAMP or IP3/DAG | Neuromodulation, pre/postsynaptic regulation |
- Inactivation: Reuptake via EAAT (Excitatory Amino Acid Transporters, SLC1 family) into presynaptic neurons AND astrocytes. In astrocytes, glutamate is converted to glutamine by glutamine synthetase → glutamine exported back to neurons (glutamate-glutamine cycle).
- Excitotoxicity: Excess synaptic glutamate (from energy failure, ischemia, hypoglycemia) causes prolonged NMDA receptor activation → massive Ca2+ influx → activation of destructive enzymes (calpains, endonucleases, nitric oxide synthase) → neuronal death. Key in stroke, TBI, hypoglycemia, and neurodegenerative diseases.
B. Aspartate
- Excitatory NT, but functions in fewer pathways than glutamate.
- Synthesized from the TCA cycle intermediate oxaloacetate via transamination.
- Acts on NMDA receptors (co-agonist with glutamate).
- Cannot cross the blood-brain barrier.
C. Acetylcholine (Excitatory at NMJ and ANS)
At the neuromuscular junction (NMJ) and in many ANS synapses, ACh is excitatory through nicotinic receptors (ionotropic, Na+/K+ channels → depolarization).
General Characteristics of Excitatory Neurotransmitters
- Depolarize the postsynaptic membrane - move resting membrane potential toward the action potential threshold (more positive = more likely to fire)
- Act on ionotropic receptors (fast, direct ion channel gating) or Gq-coupled metabotropic receptors (slow, via second messengers)
- Increase intracellular Ca2+ (especially via NMDA) - critical for synaptic plasticity and gene expression
- Terminated by reuptake (via EAATs for glutamate/aspartate) or hydrolysis (ACh via AChE)
- Dual role: physiologic excitation is essential for all brain function; pathological excess causes excitotoxic neuronal death
Sources: - Basic Medical Biochemistry 6e, p. 1648-1851 | Guyton & Hall, p. 750-851
7. Function and Properties of Endocannabinoids
What Are Endocannabinoids?
Endocannabinoids are endogenous lipid-derived messengers that activate cannabinoid receptors. Unlike classical neurotransmitters, they are:
- Synthesized on demand (not stored in vesicles) from membrane phospholipid precursors
- Released from the postsynaptic neuron (not presynaptic)
- Act in a retrograde direction - traveling backward across the synapse to inhibit the presynaptic terminal
Major Endocannabinoids
| Compound | Full Name | Precursor | Synthesizing Enzyme | Degrading Enzyme |
|---|---|---|---|---|
| AEA (Anandamide) | N-arachidonoylethanolamine | Phosphatidylethanolamine | NAPE-PLD (N-acyl phosphatidylethanolamine phospholipase D) | FAAH (Fatty Acid Amide Hydrolase) |
| 2-AG | 2-arachidonoylglycerol | Diacylglycerol | DAG lipase (DAGL-α, DAGL-β) | MAGL (Monoacylglycerol Lipase) |
Receptors
| Receptor | Location | G-Protein | Effect |
|---|---|---|---|
| CB1 | Brain (especially hippocampus, cerebral cortex, basal ganglia, cerebellum), spinal cord, peripheral nerves | Gi | ↓cAMP, ↓Ca2+ channels, ↑K+ channels → inhibits presynaptic neurotransmitter release |
| CB2 | Immune cells (spleen, tonsils, microglia), periphery | Gi | Immunomodulation, inflammation |
Mechanism of Retrograde Signaling
- Postsynaptic neuron is strongly depolarized (e.g., by glutamate) → Ca2+ enters → activates DAG lipase → 2-AG is synthesized from membrane DAG
- 2-AG (or AEA) is released backward across the synapse
- Binds to CB1 receptors on the presynaptic terminal
- Gi activation → ↓cAMP and inhibition of voltage-gated Ca2+ channels → reduced Ca2+-dependent neurotransmitter vesicle release
- Net effect: depolarization-induced suppression of inhibition (DSI) or excitation (DSE) → negative feedback control of synaptic strength
Key Functions
| Function | Detail |
|---|---|
| Synaptic plasticity | Involved in LTD (long-term depression); retrograde signaling fine-tunes synaptic strength |
| Pain modulation | CB1 activation in spinal cord and brain → analgesia |
| Appetite regulation | Hypothalamic CB1 stimulation → increases appetite (explains "munchies" from cannabis) |
| Memory and learning | Hippocampal EC signaling is involved in working memory and extinction of fear memories |
| Neuroprotection | Anti-inflammatory via CB2; reduce glutamate excitotoxicity |
| Mood regulation | CB1 modulates anxiety and stress responses |
| Motor control | High CB1 density in basal ganglia and cerebellum → modulates movement |
Properties Summary
- Lipid messengers (not water-soluble like peptides or amino acids)
- Synthesized on demand from arachidonic acid-containing phospholipids
- Retrograde messengers - unique among neurotransmitters
- Short half-life due to rapid degradation by FAAH (for AEA) and MAGL (for 2-AG)
- Pharmacological targets: THC (cannabis) is a partial CB1/CB2 agonist; FAAH inhibitors (under development) increase anandamide levels for pain/anxiety
Sources: - Kaplan & Sadock's Comprehensive Textbook of Psychiatry (Peters et al., 2021 reference)
8. Mechanism and Symptoms of Parkinson's Disease
Core Pathological Mechanism
Parkinson's disease (PD) is fundamentally caused by progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) of the basal ganglia, resulting in dopamine depletion in the striatum (caudate + putamen).
Biochemical Mechanisms
A. Dopamine Pathway Disruption
- The nigrostriatal dopamine pathway (SNpc → striatum) normally facilitates motor control by modulating the direct and indirect pathways of the basal ganglia.
- Loss of >60-80% of SNpc neurons and >80% of striatal dopamine is needed for symptoms to appear.
- Loss of dopamine disrupts the balance between the direct pathway (Go signal - normally facilitated by D1 receptors) and indirect pathway (Stop signal - normally inhibited by D2 receptors) → net overactivity of the subthalamic nucleus and GPi/SNpr → excessive inhibition of thalamus → reduced motor cortex activation
B. Alpha-Synuclein and Lewy Bodies
- The hallmark pathological finding is Lewy bodies - intraneuronal inclusions composed primarily of alpha-synuclein (misfolded and aggregated).
- Normally, alpha-synuclein is involved in vesicle cycling and dopamine release. When misfolded, it forms toxic oligomers → disrupts mitochondrial function, impairs proteasomal and lysosomal degradation → neuronal death.
- Mutations in SNCA (alpha-synuclein gene), LRRK2, Parkin, PINK1, and DJ-1 genes cause familial PD.
C. Mitochondrial Dysfunction & Oxidative Stress
- Dopaminergic neurons are especially vulnerable because dopamine metabolism via MAO generates H2O2 (hydrogen peroxide) → oxidative stress.
- Mitochondrial complex I deficiency is found in SNpc of PD patients → reduced ATP + increased reactive oxygen species → neuronal death.
- The neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is a classic experimental model of PD: MPTP is converted to MPP+ by MAO-B → enters dopaminergic neurons via DAT → inhibits complex I → oxidative damage → selective SNpc neuron death.
D. Neuroinflammation
- Activated microglia surround Lewy body-containing neurons → release cytokines and reactive oxygen species → amplify neurodegeneration.
Symptoms
The hallmark motor symptoms arise from dopamine depletion in the striatum:
| Symptom | Mechanism |
|---|---|
| Resting tremor (3-6 Hz, "pill-rolling") | Imbalance between dopamine and acetylcholine in striatum; diminishes with movement |
| Rigidity (cogwheel or lead-pipe) | Increased muscle tone due to disinhibited stretch reflexes and altered basal ganglia output |
| Bradykinesia (slowness of movement) | Reduced motor cortex activation from overactive GPi inhibition of thalamus |
| Postural instability | Loss of postural reflexes; leads to falls |
| Shuffling gait, hypomimia (masked face) | Motor hypofunction |
| Micrographia | Small handwriting due to bradykinesia |
Non-motor symptoms (from involvement of other brain regions):
- Anosmia (loss of smell) - often the first symptom, due to early pathology in olfactory bulb
- REM sleep behavior disorder - acting out dreams (early sign)
- Constipation - enteric nervous system involvement
- Depression and anxiety - dopamine and serotonin deficits
- Cognitive decline/dementia (in advanced PD) - cholinergic neuron loss
- Autonomic dysfunction - orthostatic hypotension, hypersalivation
Treatment Targeting the Biochemical Defect
| Drug | Target | Mechanism |
|---|---|---|
| L-DOPA + carbidopa | Replenish dopamine | L-DOPA crosses BBB, converted to DA by AADC; carbidopa inhibits peripheral AADC |
| MAO-B inhibitors (selegiline, rasagiline) | Reduce DA breakdown | Irreversible MAO-B inhibitors; increase DA t1/2 |
| COMT inhibitors (entacapone, tolcapone) | Reduce L-DOPA/DA degradation | Extend effect of L-DOPA |
| DA agonists (pramipexole, ropinirole) | Stimulate D2/D3 receptors | Bypass presynaptic neurons |
| Anticholinergics (benztropine) | Reduce ACh dominance | Restore DA/ACh balance in striatum; best for tremor |
Sources: - Lippincott Pharmacology 6e, p. 2260-2268 | Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 157-192 | Basic Medical Biochemistry 6e
Quick Reference Summary Table
| Topic | Core Concept | Key Enzyme/Protein | Clinical Pearl |
|---|---|---|---|
| Catecholamine biosynthesis | Tyrosine → DOPA → DA → NE → Epi | TH (rate-limiting), AADC, DBH, PNMT | L-DOPA bypasses TH for Parkinson's |
| Histamine action | H1/H2/H3/H4 receptors | HNMT (brain), DAO (periphery) | Scombroid poisoning - excess histamine from fish |
| Peptide NT inactivation | Extracellular proteolysis, no reuptake | Neprilysin, DPPIV, ACE, aminopeptidases | ACE inhibitors / DPP-IV inhibitors used clinically |
| Alzheimer's disease | Aβ plaques + tau tangles + ACh loss | APP, BACE1, γ-secretase, AChE | AChEI (donepezil) + memantine (NMDA block) |
| Catecholamine transporters | DAT, NET, SERT (plasma); VMAT2 (vesicle) | SLC6 family (plasma), SLC18 family (vesicle) | Cocaine blocks all three; amphetamine reverses DAT |
| Excitatory neurotransmitters | Depolarization; Ca2+ influx | AMPA/NMDA/kainate receptors; EAAT | Glutamate excitotoxicity in stroke/hypoglycemia |
| Endocannabinoids | Retrograde, lipid-based, on-demand | FAAH (degrades AEA), MAGL (degrades 2-AG) | CB1 retrograde inhibition; appetite, pain, memory |
| Parkinson's disease | SNpc DA neuron loss → striatum DA deficit | Alpha-synuclein, LRRK2, Complex I (mitoch) | Tremor + rigidity + bradykinesia; treat with L-DOPA |
Good luck on your exam! Each answer here maps directly to the core biochemical mechanisms your curriculum covers.
This is a shared conversation. Sign in to Orris to start your own chat.