Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have everything needed for a thorough, well-sourced answer. Here is the full explanation:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
Overview
ARDS is a syndrome of acute hypoxemic respiratory failure caused by diffuse alveolar damage (DAD). The unifying mechanism is a breakdown of the alveolar-capillary barrier - whether triggered by a direct lung insult (pneumonia, aspiration, inhalation injury) or an indirect/systemic insult (sepsis, trauma, pancreatitis, transfusions) - leading to non-cardiogenic pulmonary edema, surfactant failure, and intractable hypoxia.
Berlin Criteria (2012, updated 2023): PaO2/FiO2 ratio ≤300 mmHg on ≥5 cmH2O PEEP, bilateral opacities on imaging within 1 week of a known clinical insult, not fully explained by cardiac failure.
The Alveolar-Capillary Barrier Under Normal Conditions
The alveolus is protected by two cell populations:
- Type I pneumocytes (alveolar epithelium) - cover ~95% of the alveolar surface; thin gas-exchange cells
- Type II pneumocytes - produce surfactant; serve as progenitors for type I cells
- Pulmonary microvascular endothelium - sits on the capillary side, normally forms a tight barrier
These two layers together form the alveolar-capillary membrane. Loss of integrity on either side is necessary and sufficient for ARDS to develop. - Murray & Nadel's Textbook of Respiratory Medicine, p. 3083
Initiating Events: Two Pathways of Injury
Direct (Pulmonary) Insult
Pneumonia, aspiration of gastric contents, toxic inhalation, pulmonary contusion, and near-drowning injure pneumocytes directly. Damaged type I cells release damage-associated molecular patterns (DAMPs) sensed by resident alveolar macrophages, which then secrete TNF-α and IL-1β, acting on neighboring endothelium.
Indirect (Extrapulmonary) Insult
Sepsis, severe trauma, pancreatitis, and massive transfusions generate circulating inflammatory mediators - bacterial lipopolysaccharide (LPS), TNF-α, IL-1, IL-6, IL-8 - that activate pulmonary endothelial cells directly via the bloodstream.
In both cases, the downstream events are virtually identical. - Robbins, Cotran & Kumar Pathologic Basis of Disease, p. 631
Core Pathogenic Mechanism: Step-by-Step
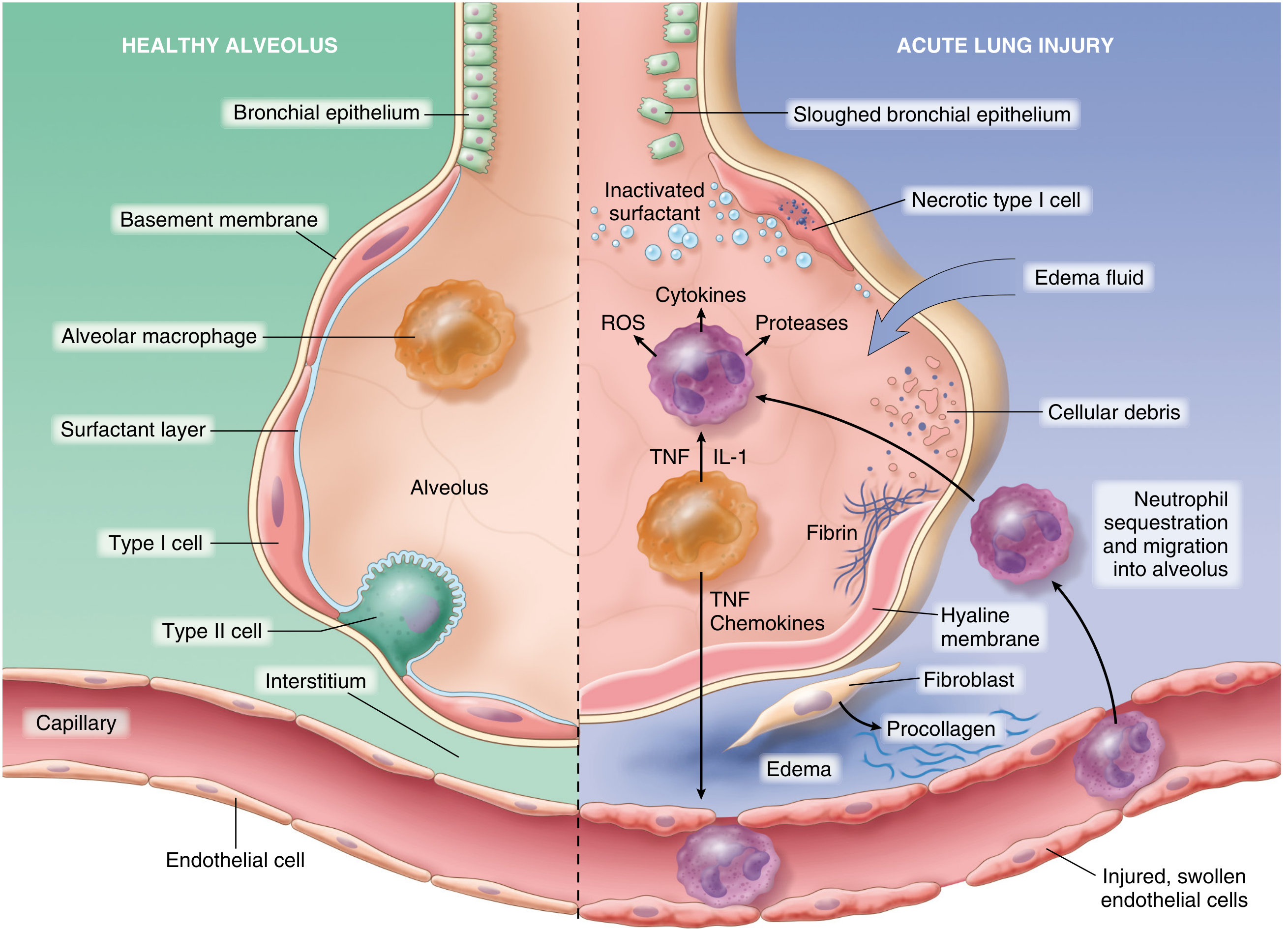
Step 1 - Endothelial Activation
Injury or circulating mediators cause pulmonary endothelial cells to upregulate:
- Adhesion molecules (ICAM-1, E-selectin, P-selectin)
- Procoagulant proteins (tissue factor, PAI-1)
- Chemokines (notably IL-8/CXCL8)
This primes the endothelium to capture circulating neutrophils.
Step 2 - Neutrophil Sequestration and Transmigration
Neutrophils adhere to activated endothelium and migrate into the interstitium and alveoli. This is the central amplification step. Once inside the alveolus, neutrophils degranulate and release a destructive arsenal:
| Mediator | Mechanism of injury |
|---|---|
| Neutrophil elastase (NE) | Degrades epithelial/endothelial cadherins (adherens junctions), disrupts barrier integrity; also degrades growth factors and cytokines |
| Matrix metalloproteinases (MMPs) | Break down junctional proteins in epithelium and endothelium, induce cell death |
| Reactive oxygen species (ROS) | Oxidative damage to cell membranes, disruption of tight junctions, potentiation of proteinase injury |
| Reactive nitrogen species | Similar oxidative/nitrosative cell death |
| Neutrophil extracellular traps (NETs) | Web-like DNA/histone structures that trap pathogens but also damage endothelium, promote thrombus formation, and amplify IL-6/TNF production |
| Phospholipase A2 | Degrades surfactant phospholipids, promoting alveolar collapse |
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 3151-3157
Important caveat: ARDS can develop even in severely neutropenic patients, indicating that alveolar macrophages, monocytes, platelets, and circulating mediators can drive injury independently of neutrophils.
Step 3 - Barrier Disruption and Protein-Rich Edema
Endothelial and epithelial injury produce:
- Increased microvascular permeability - protein-rich plasma fluid (including albumin, fibrinogen) floods the interstitium and alveolar spaces
- Loss of alveolar fluid clearance - type I pneumocyte necrosis prevents active sodium/water reabsorption
- Surfactant dysfunction - type II pneumocyte death plus dilution/inactivation by edema fluid removes the surface tension-lowering layer, causing alveolar collapse (atelectasis)
The net result is non-cardiogenic pulmonary edema with a PAWP <18 mmHg (distinguishing it from cardiogenic causes).
Step 4 - Coagulation and Microvascular Occlusion
Simultaneously, vascular injury triggers:
- Microthrombus formation in pulmonary capillaries (via tissue factor/thrombin axis)
- Fibrocellular proliferation in vessel walls
- Platelet-neutrophil aggregates further amplifying vascular permeability
This obliterates portions of the pulmonary vasculature, redirecting blood flow and increasing pulmonary dead space and pulmonary vascular resistance, contributing to pulmonary hypertension and right heart strain. - Harrison's Principles of Internal Medicine 22E, p. 2654
Step 5 - Hyaline Membrane Formation
Condensed plasma proteins, cellular debris, and dysfunctional surfactant components organize in the air spaces into the characteristic hyaline membranes - eosinophilic, glassy deposits lining the alveolar walls. These are the histologic hallmark of DAD.
Pathophysiologic Consequences (Why Patients Deteriorate)
The above cellular events produce these physiologic derangements:
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + atelectasis in dependent zones | Intrapulmonary shunt - blood perfuses non-ventilated alveoli; hypoxemia refractory to supplemental O2 |
| Heterogeneous compliance loss | Stiff ("baby") lungs requiring high driving pressures |
| Microvascular occlusion | Increased dead space + pulmonary hypertension |
| Surfactant loss | Further atelectasis, reduced compliance |
| Increased work of breathing | Respiratory muscle fatigue, respiratory failure |
The hypoxemia in ARDS is characteristically refractory - it does not respond well to FiO2 alone because shunted blood bypasses ventilated regions entirely. - Harrison's, p. 2654
The Three Phases of ARDS

Phase 1 - Exudative (Days 0-7)
- Alveolar capillary injury, protein-rich edema, neutrophilic infiltration
- Hyaline membrane formation
- Surfactant dysfunction and alveolar collapse
- Severe hypoxemia and decreased compliance
- Clinical onset typically 12-36 hours after the precipitating insult (occasionally delayed up to 5-7 days)
Phase 2 - Proliferative (Days 7-21)
- Hyaline membranes reorganize
- Interstitial inflammation with shift from neutrophil- to lymphocyte-predominant infiltrate
- Type II pneumocytes proliferate along alveolar basement membranes, synthesizing new surfactant and differentiating into type I cells
- Early fibrosis and collagen deposition appear
- N-terminal procollagen peptide III (a collagen synthesis marker) can be detected in BAL fluid as early as 24 hours after onset, suggesting fibroproliferation begins far earlier than previously thought
- Murray & Nadel's, p. 3074-3076
Phase 3 - Fibrotic (>21 days, subset of patients)
- Alveolar edema and inflammatory exudates convert to extensive alveolar-duct and interstitial fibrosis
- Emphysema-like changes with large bullae from disrupted acinar architecture
- Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and pulmonary hypertension
- Increased risk of pneumothorax, further reduced compliance, increased dead space
- Lung biopsy evidence of fibrosis at any phase of ARDS independently predicts increased mortality
- Harrison's, p. 2694
Ventilator-Induced Lung Injury (VILI): A Secondary Mechanism
ARDS lungs are heterogeneous - some regions are consolidated and fluid-filled, others are relatively normal. Because the normal regions bear disproportionate tidal volume, standard ventilation produces:
- Volutrauma/Barotrauma - overdistention of relatively spared alveoli at high tidal volumes
- Atelectrauma - repetitive collapse-reopening of unstable alveoli
- Biotrauma - mechanical stretch triggers cytokine release, perpetuating systemic inflammation and multi-organ failure
This is the rationale for lung-protective ventilation: tidal volumes of 6 mL/kg predicted body weight, plateau pressures ≤30 cmH2O (ARDS Network trial). - Goldman-Cecil Medicine, p. 1066
Mediator Summary
| Category | Key Players | Effect |
|---|---|---|
| Proinflammatory cytokines | TNF-α, IL-1β, IL-6, IL-8 | Endothelial activation, neutrophil recruitment, systemic inflammation |
| Lipid mediators | Leukotriene B4, platelet-activating factor | Neutrophil chemotaxis, vascular permeability |
| Proteases | Neutrophil elastase, MMP-2, MMP-9 | Junctional protein degradation, cell death |
| Oxidants | ROS, reactive nitrogen species | Membrane injury, tight junction disruption, antiprotease inactivation |
| NETs | DNA/histones/MPO/NE | Endothelial damage, thrombosis, cytokine amplification |
| Resolution mediators | TGF-β, PDGF (late) | Fibroblast activation, collagen deposition, scar formation |
Resolution
In patients who recover, macrophages clear intraalveolar debris and, critically, switch from a pro-inflammatory to a pro-resolution phenotype. Surviving type II pneumocytes proliferate to repopulate the alveolar lining. Uninjured capillary endothelium proliferates to restore vascular integrity. The edema is reabsorbed via active sodium transport (primarily through ENaC channels on type II cells). This process may take weeks to months, and long-term pulmonary function impairment is common even in survivors. - Robbins & Cotran, p. 631
Sources:
- Harrison's Principles of Internal Medicine, 22E (2025, McGraw Hill), Ch. 312, pp. 2641-2694
- Murray & Nadel's Textbook of Respiratory Medicine, Ch. 134, pp. 3072-3157
- Robbins, Cotran & Kumar: Pathologic Basis of Disease, Ch. 15, pp. 631-632
- Goldman-Cecil Medicine, Ch. 91, pp. 1065-1067
This is a shared conversation. Sign in to Orris to start your own chat.