Nephrotic syndrome According to nephro standard textbook
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have rich, comprehensive content from the nephrology textbooks. Let me compile the full answer.
Nephrotic Syndrome
Sources: Comprehensive Clinical Nephrology, 7th Ed. | Brenner and Rector's The Kidney | NKF Primer on Kidney Diseases, 8th Ed.
Definition
Nephrotic syndrome is pathognomonic of glomerular disease. It is defined as a clinical pentad of:
- Proteinuria > 3.5 g/24 hours (nephrotic-range)
- Hypoalbuminemia (serum albumin < 3.5 g/dL)
- Edema (pitting, often periorbital and dependent)
- Hyperlipidemia
- Lipiduria (oval fat bodies, fatty casts on urinalysis)
Not all patients with proteinuria above 3.5 g/24 h will have the full syndrome - some maintain a normal serum albumin if hepatic synthesis rises adequately in response to proteinuria.
Comprehensive Clinical Nephrology, 7th Ed., p. 239
Etiology
Primary (Idiopathic) Causes
| Cause | Adults | Children |
|---|---|---|
| Minimal Change Disease (MCD) | ~15% | 70-90% |
| Focal Segmental Glomerulosclerosis (FSGS) | Most common adult primary cause; higher in African Americans | Less common |
| Membranous Nephropathy (MN) | Common in white adults | Rare |
| Membranoproliferative GN (MPGN) | Less common | Occasional |
Secondary Causes
- Diabetic kidney disease - most common secondary cause worldwide
- Amyloidosis (AL or AA)
- Lupus nephritis (Class V membranous)
- Infections: Hepatitis B, Hepatitis C, HIV, malaria
- Drugs: NSAIDs (MCD), penicillamine, gold, heroin (FSGS)
- Malignancy: Hodgkin lymphoma (MCD), solid tumors (membranous)
The prevalence of FSGS in African Americans is increased, which explains why FSGS is becoming more common in US adults but not European adults.
Comprehensive Clinical Nephrology, 7th Ed., p. 239-240
Pathophysiology of Key Features
1. Hypoalbuminemia
Hypoalbuminemia is mainly a consequence of urinary losses. The liver responds by increasing albumin synthesis, but this compensatory mechanism is blunted in nephrotic syndrome. The net protein synthesis response is non-discriminating - large molecular weight proteins not lost in urine (fibrinogen, lipoproteins, clotting factors) actually increase in plasma, while smaller proteins (albumin, antithrombin III, protein C, IgG) fall. This explains much of the metabolic cascade of nephrotic syndrome.
Muehrcke lines (paired white transverse bands on nails) are a characteristic sign of hypoalbuminemia.
Comprehensive Clinical Nephrology, 7th Ed., p. 240
2. Edema - Underfill vs. Overfill
Two mechanisms operate:
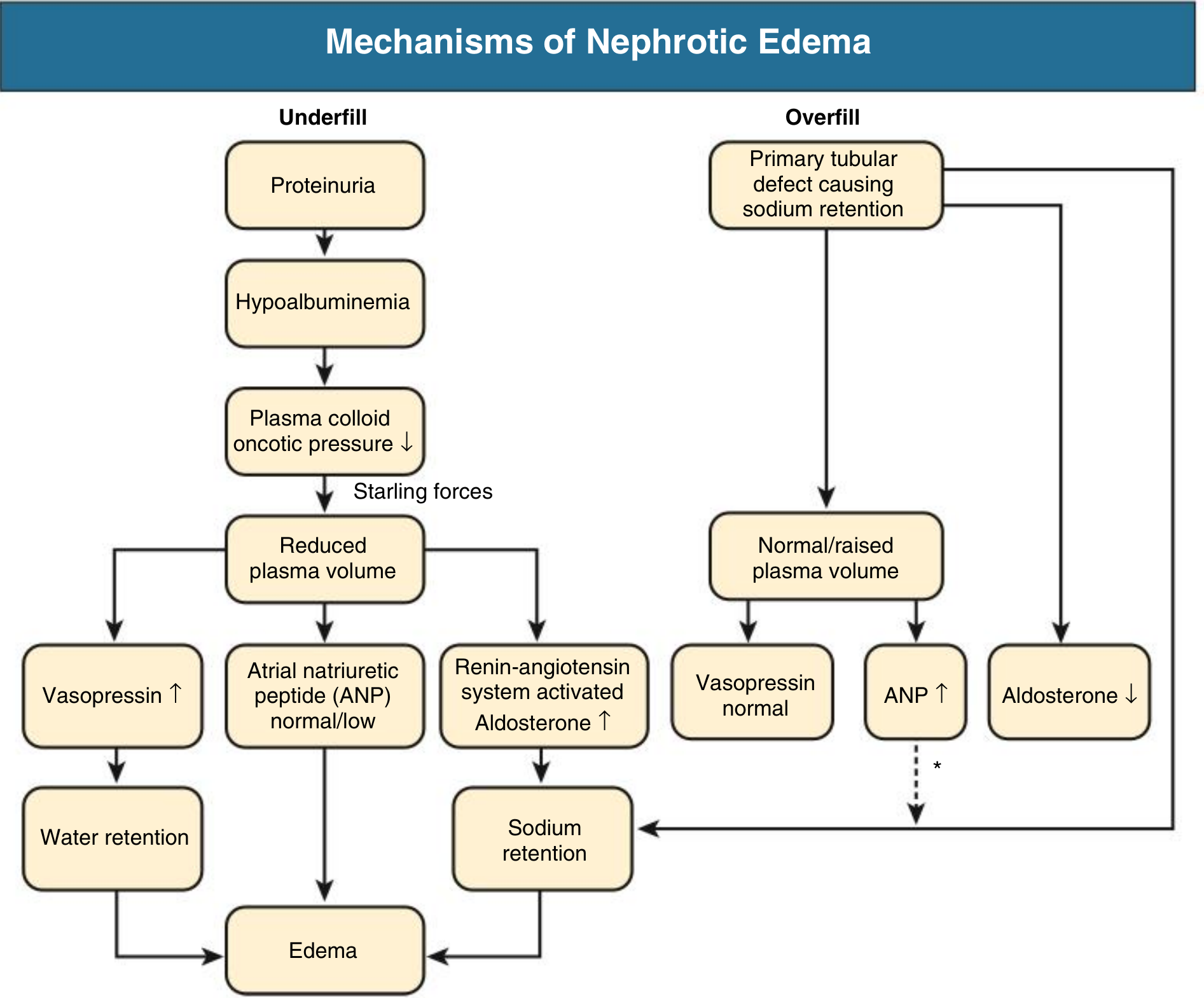
Underfill mechanism (classic, seen in MCD):
- Massive proteinuria → hypoalbuminemia → decreased plasma oncotic pressure → fluid shifts to interstitium → reduced circulating volume → RAAS activation → aldosterone-driven sodium retention → worsens edema
Overfill mechanism (more common in most other nephrotic diseases):
- Primary intrinsic renal defect in sodium excretion, involving activation of the epithelial sodium channel (ENaC) by proteolytic enzymes entering the tubular lumen during heavy proteinuria
- Results in expanded blood volume, suppressed renin/angiotensin/vasopressin, and tendency to hypertension rather than hypotension
- The kidney is relatively resistant to atrial natriuretic peptide (ANP) in this setting
Patients with MCD often have a contracted plasma volume and stimulated RAAS (underfill), whereas those with other causes of nephrotic syndrome usually have an expanded plasma volume and suppressed RAAS (overfill).
Comprehensive Clinical Nephrology, 7th Ed., p. 240; Brenner & Rector, p. 2277
3. Negative Nitrogen Balance
Heavy proteinuria leads to marked negative nitrogen balance. Nephrotic syndrome is a wasting illness, with loss of 10-20% of lean body mass possible - though this is masked by edema. Increasing protein intake does not help because the hemodynamic response is a rise in glomerular pressure, increasing urinary protein losses further. A low-protein diet reduces proteinuria, but reduces albumin synthesis rate.
4. Hypercoagulability
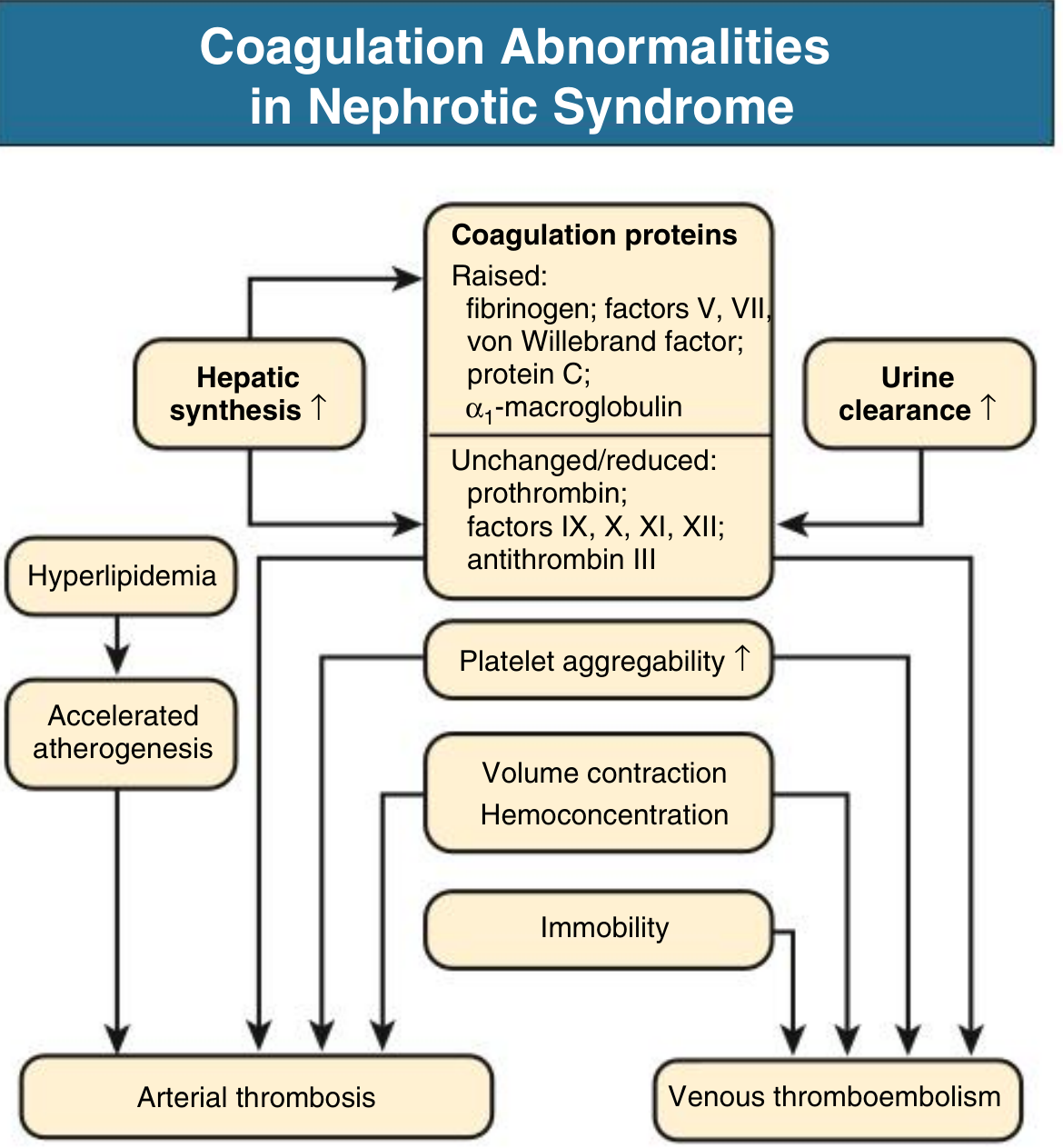
Due to altered coagulation factor levels and enhanced platelet aggregation:
- Increased (raised): Fibrinogen, Factors V and VII, von Willebrand factor, protein C, alpha-1-macroglobulin (due to increased hepatic synthesis of large proteins)
- Decreased (lost in urine): Antithrombin III, Factors IX, X, XI, XII, protein S, plasminogen
- Net effect: Hypercoagulable state, worsened by immobility, infection, and hemoconcentration
Clinical consequences:
- Up to 10% of nephrotic adults will have a clinical thromboembolic event
- Renal vein thrombosis is clinically apparent in up to 8%, and up to 10-50% when systematically sought
- Risk is particularly high in membranous nephropathy and amyloidosis
- Thromboembolic events increase greatly when serum albumin falls below 2 g/dL
- Urinary loss of protein C, protein S, plasminogen, and antithrombin III underlies the prothrombotic state
- Renal vein thrombosis can occur when proteinuria > 10 g/24 hr and albumin < 2 g/dL
Comprehensive Clinical Nephrology, 7th Ed., p. 241; NKF Primer, 8th Ed.
5. Hyperlipidemia and Lipiduria
Serum cholesterol can exceed 500 mg/dL. The lipid profile is highly atherogenic. Mechanisms include:
- Increased hepatic synthesis of LDL, VLDL, and lipoprotein(a) secondary to hypoalbuminemia
- Defective peripheral lipoprotein lipase activity → increased VLDL
- Urinary losses of HDL
- Overexpression of PCSK9 in kidney and liver (a potential therapeutic target with PCSK9 antibodies)
Nephrotic patients have approximately fivefold increased risk for coronary death - except those with MCD, as their syndrome is typically transient before remission.
Lipiduria (the 5th component) manifests as oval fat bodies and fatty casts. It results from the proteinuria itself, not the plasma lipid levels.
Comprehensive Clinical Nephrology, 7th Ed., p. 241
6. Other Metabolic Effects
- Vitamin D deficiency: Urinary loss of vitamin D-binding protein → low plasma 25-hydroxyvitamin D (however, plasma free vitamin D is usually normal; overt osteomalacia is rare)
- Iron deficiency anemia: Loss of transferrin in urine
- Hypogammaglobulinemia: Loss of IgG → susceptibility to encapsulated bacterial infections (pneumococcal peritonitis is classic)
- Hypothyroidism: Loss of thyroxine-binding globulin
- Elevated ESR: Due to hypoproteinemia and dysproteinemia - NOT a reliable marker of acute phase response in nephrotic patients
Nephrotic vs. Nephritic Syndrome
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Onset | Insidious | Abrupt |
| Edema | ++++ | ++ |
| Blood pressure | Normal | Raised |
| JVP | Normal/low | Raised |
| Proteinuria | ++++ | ++ |
| Hematuria | May/may not | +++ |
| RBC casts | Absent | Present |
| Serum albumin | Low | Normal/slightly reduced |
Comprehensive Clinical Nephrology, 7th Ed., p. 243
Complications
- Thromboembolism: DVT, PE, renal vein thrombosis, rarely arterial thrombosis
- Infections: Spontaneous bacterial peritonitis (Streptococcus pneumoniae), cellulitis
- AKI: From volume depletion, sepsis, intrarenal edema, renal vein thrombosis, adverse drug effects (NSAIDs, ACE inhibitors), or transformation to crescentic GN
- Cardiovascular disease: Accelerated atherosclerosis from sustained hyperlipidemia
- Progressive CKD: With the exception of MCD, most causes are associated with progressive kidney failure
- Risk for progression is uncommon with sustained proteinuria < 2 g/day
- Marked risk when protein excretion > 5 g/day
- Proteinuria itself is tubulointerstitially toxic
Investigations
- Urine: Dipstick proteinuria +++ or ++++; 24-hour urine protein OR spot urine protein:creatinine ratio; microscopy for oval fat bodies, fatty casts
- Blood: Serum albumin (low), serum cholesterol/triglycerides (elevated), renal function (creatinine, eGFR), FBC (anemia)
- Serology: ANA, anti-dsDNA, anti-PLA2R (membranous), complement C3/C4, hepatitis B/C, HIV, serum protein electrophoresis
- Kidney biopsy: Required in adults for all new-onset nephrotic syndrome (except clinical diabetic nephropathy)
- In children, MCD is assumed and empirical steroids are given without biopsy if the presentation is typical
Management
Non-specific/General Measures
- Dietary: Low-sodium diet (< 2 g/day); protein intake 0.8-1 g/kg/day (restriction reduces proteinuria but must balance risk of worsening negative nitrogen balance)
- Anti-proteinuric therapy: ACE inhibitor or ARB - cornerstone of management regardless of etiology (reduces intraglomerular pressure and proteinuria)
- Edema: Loop diuretics (furosemide); combination with thiazide for resistant edema; note that hypoalbuminemia reduces furosemide binding to plasma proteins and enlarges its volume of distribution, potentially impairing response
- Hyperlipidemia: Statins are the primary treatment (for cardiovascular prevention); clinical evidence for CKD-slowing by statins is inconclusive
- Anticoagulation: For those at high risk (serum albumin < 2 g/dL, membranous nephropathy, immobility); prophylactic anticoagulation is controversial but required for established thrombosis
- Infection prevention: Pneumococcal vaccination; awareness of susceptibility to encapsulated organisms
Disease-Specific Treatment
| Disease | First-line Treatment |
|---|---|
| MCD | Corticosteroids (prednisolone) - highly responsive |
| FSGS (primary) | High-dose corticosteroids; calcineurin inhibitors (tacrolimus/cyclosporine) for steroid-resistance |
| Membranous nephropathy | Conservative management first; immunosuppression (cyclophosphamide + steroids, or rituximab) for high-risk patients |
| Lupus nephritis Class V | ACE inhibitor/ARB; hydroxychloroquine; immunosuppression for progressive disease |
| Diabetic nephropathy | RAAS blockade; SGLT2 inhibitors; glycemic control |
| Amyloidosis | Treatment of underlying cause (myeloma, chronic infection) |
Comprehensive Clinical Nephrology, 7th Ed., p. 239-243; NKF Primer on Kidney Diseases, 8th Ed.
Prognosis
Heavy proteinuria at any time during follow-up substantially worsens prognosis even without frank nephrotic syndrome. The degree of proteinuria is one of the strongest predictors of progression to CKD. MCD has an excellent renal prognosis with appropriate treatment; FSGS, membranous nephropathy, and diabetic nephropathy carry greater risk for ESKD.
Comprehensive Clinical Nephrology, 7th Ed., Fig. 16.13
Key references from nephrology standard texts:
- Comprehensive Clinical Nephrology, 7th Edition - Chapter 16 (Nephrotic Syndrome)
- Brenner and Rector's The Kidney - Chapter 50 (Edema and Nephrotic Syndrome)
- NKF Primer on Kidney Diseases, 8th Edition - Chapter 16 (Glomerular Syndromes)
Same for nephritic syndrome
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have rich, comprehensive content from multiple nephrology textbooks. Let me compile the full structured answer.
Nephritic Syndrome
Sources: Comprehensive Clinical Nephrology, 7th Ed. | Brenner and Rector's The Kidney | NKF Primer on Kidney Diseases, 8th Ed.
Definition
Nephritic syndrome is defined as the presence of glomerular hematuria (dysmorphic red blood cells or RBC casts), in combination with:
- Hypertension (raised BP, raised JVP)
- Edema (less severe than nephrotic)
- Reduced GFR with or without oliguria
- Non-nephrotic-range proteinuria
The core mechanism differs fundamentally from nephrotic syndrome: in nephritic syndrome, the glomerular injury is primarily inflammatory, causing reduction in GFR, hematuria, and sodium retention - rather than a permeability defect allowing protein leak.
Comprehensive Clinical Nephrology, 7th Ed., p. 243; NKF Primer, 8th Ed.
Nephrotic vs. Nephritic - Side-by-Side
| Feature | Nephrotic | Nephritic |
|---|---|---|
| Onset | Insidious | Abrupt |
| Edema | ++++ | ++ |
| Blood pressure | Normal | Raised |
| JVP | Normal/low | Raised |
| Proteinuria | ++++ (>3.5 g/day) | ++ (sub-nephrotic) |
| Hematuria | May or may not occur | +++ (gross or microscopic) |
| RBC casts | Absent | Present |
| Serum albumin | Low | Normal/slightly reduced |
Comprehensive Clinical Nephrology, 7th Ed., Table 16.4
Etiology - Classification by Mechanism
The classification of nephritic syndrome/RPGN is etiology-based, which is the current preferred approach:
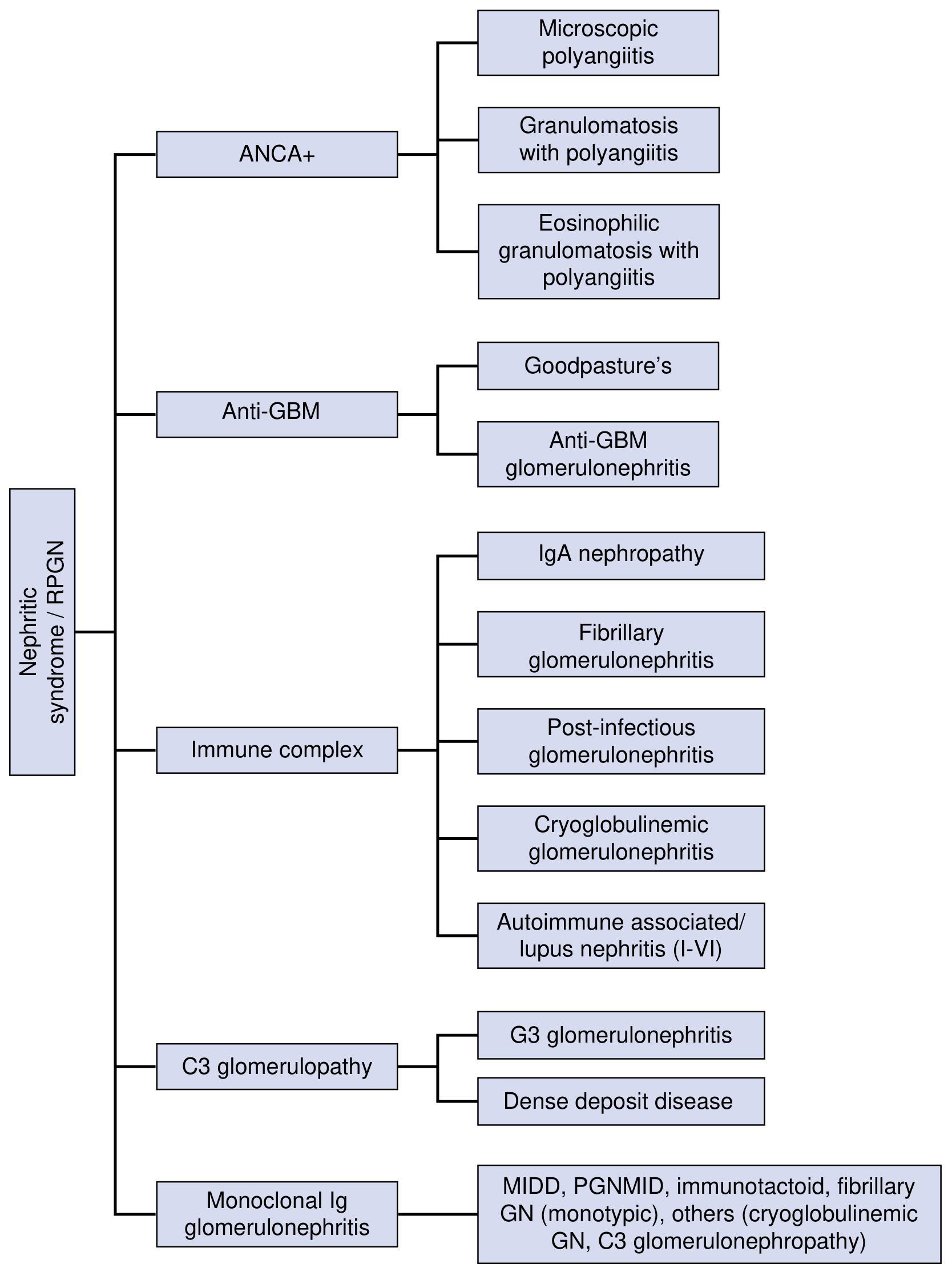
1. Immune Complex-Mediated GN
- IgA Nephropathy - most common primary glomerulopathy worldwide
- Post-infectious GN (post-streptococcal and others)
- Lupus nephritis (Classes III, IV, V)
- Cryoglobulinemic GN
- Fibrillary GN
2. Pauci-immune / ANCA-Associated GN
- Granulomatosis with Polyangiitis (GPA) - c-ANCA / anti-PR3
- Microscopic Polyangiitis (MPA) - p-ANCA / anti-MPO
- Eosinophilic Granulomatosis with Polyangiitis (EGPA)
3. Anti-GBM Antibody-Mediated
- Goodpasture Syndrome (anti-GBM + pulmonary hemorrhage)
- Anti-GBM GN (kidney only)
4. C3 Glomerulopathy (Complement-driven)
- C3 Glomerulonephritis
- Dense Deposit Disease (MPGN type II)
5. Monoclonal Immunoglobulin-Associated GN
- MIDD (Monoclonal Ig Deposition Disease), PGNMID, immunotactoid GN
NKF Primer on Kidney Diseases, 8th Ed., Fig. 16.3
Pathophysiology
In nephritic syndrome, the glomerular injury is inflammatory. Key mechanisms:
- Immune complex deposition or in-situ antibody formation activates complement, recruiting neutrophils and monocytes into glomeruli
- Endocapillary proliferation and inflammatory cell infiltration physically obstruct capillary lumens, reducing GFR
- Disruption of the GBM allows RBCs and their fragments (acanthocytes) to escape into tubular fluid, forming RBC casts in the distal nephron
- Decreased GFR triggers sodium and water retention, expanding circulating volume - leading to hypertension and volume-overload edema (raised JVP, compared to the low JVP of underfill nephrotic edema)
Brenner & Rector's The Kidney; Comprehensive Clinical Nephrology, 7th Ed.
Urinalysis - The "Nephritic Urine Sediment"
A urine sediment containing:
-
5 RBCs per high-power field
- Acanthocytes (dysmorphic RBCs - pathognomonic of glomerular bleeding)
- RBC casts or mixed RBC-WBC casts
...is characteristic of glomerular hematuria and called a nephritic urine. Hematuria causes brown/tea-colored urine (not bright red, and no clots).
Brenner & Rector's The Kidney, p. 3058
Individual Diseases Causing Nephritic Syndrome
1. Post-Infectious Glomerulonephritis (PIGN)
The classic presentation of acute nephritic syndrome. Now classified as:
- Post-infectious GN (PIGN): infection already resolved (latency period 1-4 weeks); GN develops regardless of antibiotic therapy
- Infection-associated GN: GN occurs during active, ongoing infection (e.g., infective endocarditis, staphylococcal abscess)
Classic PSGN (post-streptococcal):
- Group A beta-hemolytic streptococcal pharyngitis or skin infection
- Latency: 1-3 weeks post-pharyngitis, 3-6 weeks post-impetigo
- Presents in children: rapid onset oliguria, weight gain, generalized edema, tea-colored urine, RBC casts
- Serum albumin usually normal (proteinuria rarely nephrotic range)
- Hypertension, pulmonary edema possible - without primary cardiac disease
Serologic markers:
- Anti-streptolysin O (ASO) titer elevated
- Anti-DNAase B antibody
- Complement: C3 low, C4 normal (alternative pathway activation)
Biopsy findings:
- Light microscopy: diffuse endocapillary proliferative GN, neutrophils in capillary loops
- Immunofluorescence: granular IgG and C3 deposits in capillary walls ("starry sky")
- Electron microscopy: classic subepithelial humps (hump-shaped electron-dense deposits)
Treatment: Supportive (treat underlying infection). Prognosis better in children. Persistence of active urine sediment/GFR reduction after resolution of infection should prompt workup for alternative complement pathway abnormalities.
2. IgA Nephropathy (IgAN)
Most common primary glomerulopathy worldwide.
Pathogenesis:
- Deposition of immune complexes of anti-gliadin antibodies (IgG or IgA) and galactose-deficient IgA1 in the mesangium
- Triggered by upper respiratory or GI infections
- "Synpharyngitic hematuria" - macroscopic hematuria coinciding with (not lagging) respiratory infection
Presentations:
- Asymptomatic microscopic hematuria (most common)
- Macroscopic hematuria (synpharyngitic, episodic)
- Non-nephrotic proteinuria
- Rarely nephrotic syndrome (if coexisting MCD or with severe disease)
- Can present as IgA vasculitis (Henoch-Schönlein Purpura / HSP) with skin, joint, and intestinal involvement
Secondary IgAN associations: IBD, advanced liver disease, ankylosing spondylitis, dermatitis herpetiformis
Biopsy (Oxford/MEST-C scoring):
- M - Mesangial hypercellularity
- E - Endocapillary proliferation
- S - Segmental glomerulosclerosis
- T - Tubular atrophy/interstitial fibrosis
- C - Crescents
- Immunofluorescence: mesangial IgA deposition (defining feature)
- Electron microscopy: mesangial electron-dense deposits
Prognosis: ~60% benign course; 40% progress to ESKD over 10-20 years. Predictors of progression: renal insufficiency, hypertension, proteinuria > 1 g/24 hr at biopsy. The T score (tubular atrophy/fibrosis) is the most consistent predictor of ESKD.
Treatment: ACE inhibitor/ARB for proteinuria reduction; SGLT2 inhibitors (emerging evidence); immunosuppression (steroids, cyclophosphamide) for progressive disease; sparsentan and budesonide (targeted-release) are newer options.
3. Lupus Nephritis
- A major cause of secondary nephritic syndrome
- Classified ISN/RPS Class I-VI; Classes III (focal) and IV (diffuse) are the classic nephritic presentations
- Serologic workup: ANA, anti-dsDNA, complement C3/C4 (both low in active lupus - classical pathway)
- Treatment: Hydroxychloroquine + mycophenolate or cyclophosphamide; voclosporin/belimumab as adjuncts
4. ANCA-Associated Vasculitis (Pauci-immune GN)
- GPA (formerly Wegener's): c-ANCA (PR3-ANCA); upper/lower respiratory tract + kidney
- MPA: p-ANCA (MPO-ANCA); kidney-predominant
- EGPA (formerly Churg-Strauss): p-ANCA; asthma + eosinophilia + kidney
Biopsy: Pauci-immune necrotizing and crescentic GN (no/scant immune deposits on IF - hence "pauci-immune")
Treatment (induction): High-dose corticosteroids + rituximab (preferred) or cyclophosphamide; plasma exchange (TPE) for dialysis-dependent patients or with pulmonary hemorrhage
5. Anti-GBM Disease / Goodpasture Syndrome
- Antibodies directed against the NC1 domain of alpha-3 chain of type IV collagen in the GBM (and alveolar basement membrane)
- Goodpasture syndrome = anti-GBM GN + pulmonary hemorrhage
- Rapidly progressive; crescentic GN on biopsy
- Immunofluorescence: linear IgG deposits along GBM (pathognomonic)
- Treatment: Plasma exchange (to remove circulating anti-GBM antibodies) + cyclophosphamide + corticosteroids
- Renal outcomes depend heavily on initial creatinine: patients with Cr > 5.7 mg/dL or dialysis-dependence at presentation have very poor renal recovery (0-18%)
- Transplantation should be delayed ≥12 months after antibody levels normalize
Brenner & Rector's The Kidney, Table 66.2
Rapidly Progressive GN (RPGN)
RPGN is a clinical syndrome defined by rapid loss of kidney function over days to weeks in the context of nephritic syndrome. Untreated, it leads to ESKD.
Histologic hallmark: Crescentic GN - proliferative cellular response outside the glomerular tuft but within Bowman's space, forming a crescent shape on cross section. The glomerular tuft also shows segmental necrosis.
Three major types (Brenner & Rector classification):
- Type I - Anti-GBM disease / Goodpasture syndrome
- Type II - Immune complex-mediated (SLE, post-infectious, IgA, mixed cryoglobulinemia)
- Type III - Pauci-immune (ANCA-associated: GPA, MPA)
- Note: some patients have dual positivity (both anti-GBM and ANCA)
Pulmonary-renal syndrome (concurrent pulmonary hemorrhage + GN) occurs in:
- ANCA-associated pauci-immune GN
- Anti-GBM disease (Goodpasture)
Serologic Tests for Differential Diagnosis
| Disease | Key Serology | Complement |
|---|---|---|
| Post-streptococcal GN | ASO, anti-DNAase B | C3 low, C4 normal |
| Lupus nephritis | ANA, anti-dsDNA | C3 low, C4 low |
| IgA nephropathy | Serum IgA elevated (50%) | Normal |
| ANCA-GPA | c-ANCA / anti-PR3 | Normal |
| ANCA-MPA | p-ANCA / anti-MPO | Normal |
| Anti-GBM disease | Anti-GBM antibody | Normal |
| Cryoglobulinemia | Cryoglobulins, HCV | C3 low, C4 very low |
| C3 glomerulopathy | C3 nephritic factor | C3 low, C4 normal |
| Infective endocarditis | Blood cultures | C3 low |
Comprehensive Clinical Nephrology, 7th Ed., Table 16.5
Investigations Summary
- Urinalysis with microscopy: Hematuria, dysmorphic RBCs, RBC casts, subnephrotic proteinuria
- Urine protein:creatinine ratio (to quantify proteinuria)
- Renal function: Creatinine, eGFR (often acutely elevated in RPGN)
- Serology panel: ANA, anti-dsDNA, ANCA (PR3 + MPO), anti-GBM, complement (C3, C4), serum IgA, cryoglobulins, hepatitis B/C, HIV, blood cultures if endocarditis suspected
- Imaging: Renal ultrasound (kidneys often normal size or enlarged in acute GN)
- Kidney biopsy: Required in most cases to establish diagnosis, guide immunosuppressive treatment, and assess prognosis
Management Principles
Non-specific
- Blood pressure control: ACE inhibitors/ARBs (also anti-proteinuric)
- Fluid/sodium management: Diuretics for volume overload
- Dialysis: If RPGN progresses to oliguric renal failure
Disease-Specific
| Disease | Induction | Maintenance |
|---|---|---|
| PSGN/PIGN | Supportive; treat infection | None needed (self-limited in children) |
| IgA nephropathy | ACE inhibitor/ARB; steroids if progressive | ACEi/ARB long-term |
| Lupus (III/IV) | MMF + steroids, or cyclophosphamide + steroids | MMF or azathioprine |
| ANCA vasculitis | Rituximab or cyclophosphamide + steroids; TPE if dialysis-dependent or pulmonary hemorrhage | Rituximab or azathioprine |
| Anti-GBM | Plasma exchange + cyclophosphamide + steroids | Cyclophosphamide |
Plasma Exchange (TPE) in RPGN:
- Evidence supports TPE for anti-GBM disease and ANCA-vasculitis when dialysis-dependent or with pulmonary hemorrhage
- In ANCA-associated disease with Cr > 5.7 mg/dL: TPE reduced progression to ESKD at 1 year by 24% vs. IV methylprednisolone alone
- TPE is considered the best adjunct to immunosuppression for advanced renal disease in these contexts
- Limited evidence for use in less severe kidney disease
Brenner & Rector's The Kidney, pp. 2830-2831
Prognosis
- PSGN in children: Generally excellent - full recovery is common
- PSGN in adults: Less favorable; some develop chronic kidney disease
- IgA nephropathy: ~40% progress to ESKD over 10-20 years
- Anti-GBM with creatinine > 5.7 mg/dL at presentation: Recovery of independent renal function is very low (0-18% across studies)
- ANCA vasculitis: With early aggressive treatment, ~60-70% may avoid dialysis
- Lupus nephritis: Outcome depends on WHO/ISN class and treatment response
Key references:
- Comprehensive Clinical Nephrology, 7th Edition - Chapter 16 (Nephritic Syndrome, RPGN)
- Brenner and Rector's The Kidney - Chapter 37 & Chapter 66 (RPGN; Anti-GBM disease)
- NKF Primer on Kidney Diseases, 8th Edition - Chapter 16 (Glomerular Syndromes)
This is a shared conversation. Sign in to Orris to start your own chat.