Glomerulonephritis
Reading File
Finding Sources
Searching PubMed
glomerulonephritis classification management
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
I now have comprehensive information to write a thorough answer on glomerulonephritis. Let me compile everything:
Glomerulonephritis (GN)
Glomerulonephritis refers to a group of inflammatory and immunologic disorders that primarily injure the glomerular capillaries, leading to impaired filtration, hematuria, proteinuria, and progressive renal failure. Classification has historically been challenging due to overlapping features between infection-related and immune-mediated pathology.
Definition and General Features
GN is associated with:
- Hypertension and volume expansion (edema, orthopnea, dyspnea)
- Abnormal urinalysis - the hallmark is the "active nephritic sediment"
- Oliguria or anuria with low urine sodium and concentrated urine (Na+ and water retention)
- In children: ascites; in the elderly: restlessness and incipient pulmonary edema
The disease may present acutely (over days to weeks, as in postinfectious GN) or as a chronic smoldering process with inexorable progression over months to years. The pathologic correlate is the relative proportion of acute inflammatory/proliferative/necrotizing lesions versus chronic sclerotic and atrophic findings on renal biopsy.
Nephritic Urinalysis
The nephritic urinalysis shows:
- Hematuria - red blood cells become misshapen (dysmorphic) as they cross the damaged glomerular barrier
- Proteinuria
- Cellular casts (red cell casts, white cell casts)
- Mixed pyuria (PMN leukocytes)
- Macroscopic hematuria - "cola" or "tea-colored" urine (hemoglobin + acid urine = hematin)
Microscopic hematuria is invisible to the naked eye, but as little as 10-20 mL of blood per liter turns urine visibly red.
Pathogenesis - Mechanisms of Glomerular Injury
Three major immunologic mechanisms are recognized (Henry's Clinical Diagnosis and Management):
| Mechanism | Example |
|---|---|
| Circulating antigen-antibody immune complex deposition + complement activation | Cryoglobulinemia, SLE |
| Deposition of "planted" exogenous antigens in glomeruli, followed by antibody binding | Postinfectious GN |
| Autoantibodies to intrinsic in-situ glomerular antigens | Membranous nephropathy (anti-PLA2R) |
Additionally, ANCA (antineutrophil cytoplasmic antibodies) cause pauci-immune GN without significant immunoglobulin deposition, and cell-mediated immunity via T lymphocytes contributes to podocyte injury.
Classification by Pathology and Mechanism
Nephrotic Presentations
| Disease | Pathogenesis | LM | IF | EM |
|---|---|---|---|---|
| Membranous nephropathy | In situ immune complex (anti-PLA2R) | Capillary wall thickening | Granular IgG + C3 (capillary wall) | Subepithelial deposits |
| IgA nephropathy | Abnormal IgA glycosylation (galactose-deficient IgA1) | Mesangial hypercellularity | Mesangial dominant IgA + C3 | Mesangial deposits |
| MPGN | Immune complex deposition | Endocapillary hypercellularity, mesangial sclerosis, "double contouring" | Capillary wall IgG + C3 | Subendothelial deposits |
| C3 glomerulonephritis | Dysregulation of alternative complement pathway | Similar to MPGN | Dominant C3, minimal/no Ig | Subendothelial deposits |
| Dense deposit disease (DDD) | Dysregulation of alternative complement pathway | Similar to MPGN | Dominant C3 | Dense deposits within GBM |
Nephritic Presentations
| Disease | Pathogenesis | LM | IF | EM |
|---|---|---|---|---|
| Postinfectious GN | Circulating/planted immune complexes + complement | Endocapillary hypercellularity | Capillary wall IgG + C3 | Subepithelial "humps" |
| Anti-GBM disease (Goodpasture) | IgG autoAb against α3 chain of type IV collagen | Capillary wall necrosis + crescents | Linear IgG + C3 | No deposits; GBM ruptures |
| Pauci-immune GN (ANCA) | ANCA (anti-PR3 or anti-MPO) | Capillary wall necrosis + crescents | No or sparse Ig/C3 | No or sparse deposits |
| Lupus nephritis (class I-VI) | Immune complex deposition | Variable (class-dependent) | "Full-house" (IgG, IgM, IgA, C3, C1q) | Numerous deposits |
(Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 1249)
Major Specific Types
1. Postinfectious GN (PIGN)
The classic example of acute GN. Immune response involves formation of antibody-bacterial antigen complexes, typically 10 days to 3 weeks after infection by nephritogenic strains of beta-hemolytic Group A streptococci (pharynx or skin/impetigo).
Key features:
- Immune complexes activate complement cascade → intense inflammation → low C3 in serum
- Proliferation causes "bloodless capillaries" → oliguria/anuria
- Evidence of prior infection: ASO titer, anti-DNase B, streptozyme
- Subepithelial "humps" on EM
- Over time, subepithelial deposits develop (humps) and proteinuria rises; as subendothelial deposits clear, nephritic signs resolve
PIGN vs. IgA nephropathy distinction: PIGN occurs 10-21 days post-pharyngitis; IgAN hematuria occurs simultaneously with or 1-3 days into a viral pharyngitis (synpharyngitic). Only PIGN causes low serum C3.
(Harrison's Principles of Internal Medicine 22E, p. 2398)
2. IgA Nephropathy (Berger Disease)
One of the leading causes of renal failure worldwide. Caused by mesangial deposition of C3 binding to excess galactose-deficient IgA-1.
- Common in Asia; gross hematuria during upper respiratory tract infections
- C3 deposited in mesangium (not capillary loops) - serum C3 remains normal
- Management: control hypertension and proteinuria with ACE inhibitors/ARBs
- Recent advances: targeted complement pathway therapies based on biopsy findings
- RPGN form: treat with corticosteroids + cyclophosphamide
3. Rapidly Progressive GN (RPGN / Crescentic GN)
A clinical syndrome defined by severe glomerular injury with crescents (>50% of glomeruli). Three immunopathologic categories:
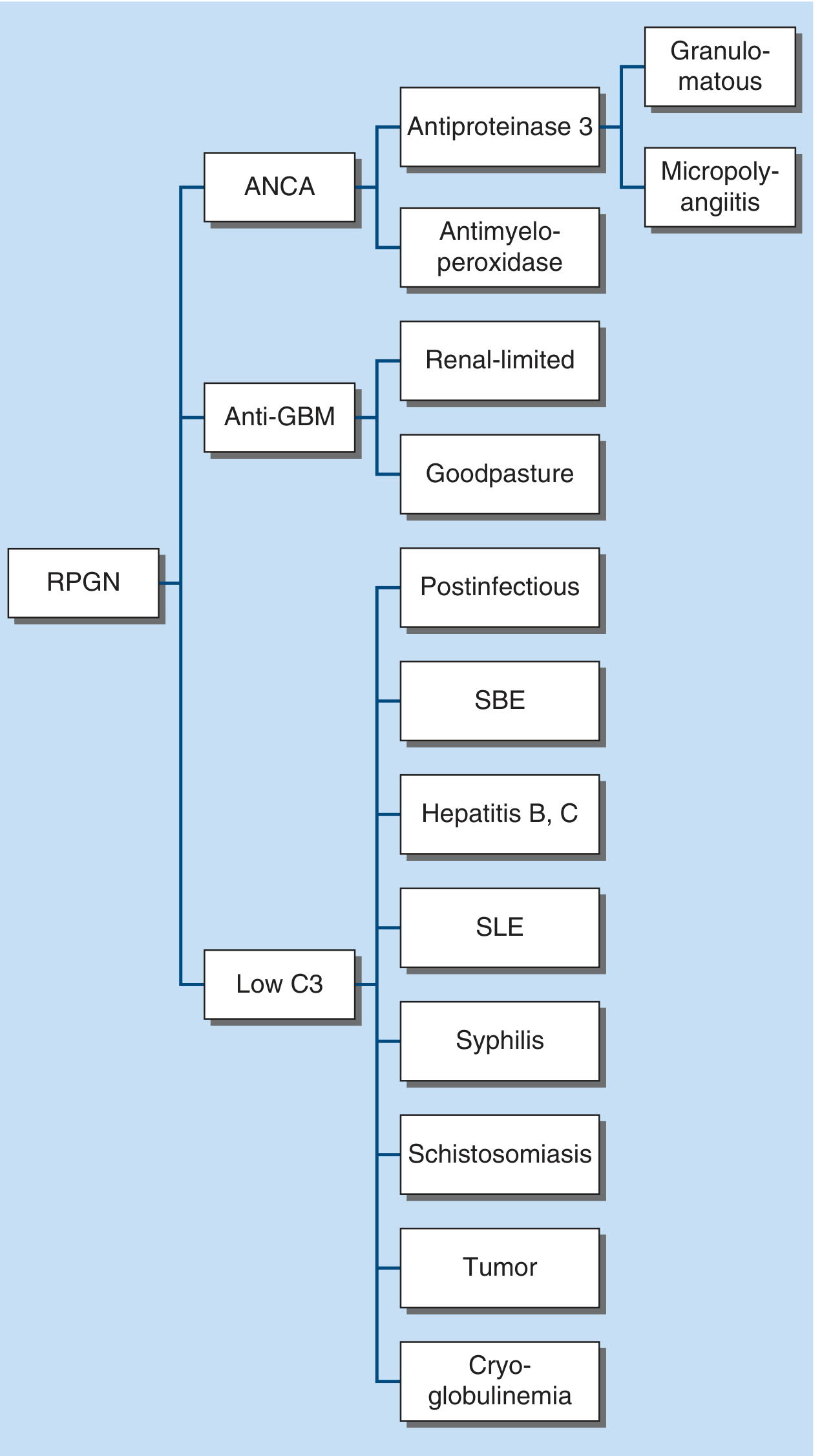
Category 1 - ANCA-positive (Pauci-immune)
- Anti-PR3 (antiproteinase-3) → Granulomatosis with polyangiitis (GPA)
- Anti-MPO (antimyeloperoxidase) → Microscopic polyangiitis (MPA)
- Most common type overall; most common in adults >60 years
- Pauci-immune: little/no Ig staining on IF
Category 2 - Anti-GBM antibody
- Renal-limited: anti-GBM GN only
- Goodpasture syndrome: anti-GBM + pulmonary hemorrhage
- IgG autoAb directed at α3 chain of NC1 domain of type IV collagen (absent in hereditary nephritis/Alport syndrome - patients with Alport receiving a transplant may develop fulminant anti-GBM disease)
- Linear IgG on IF
- Treatment: plasmapheresis to remove antibody (may take weeks-months)
Category 3 - Immune complex (Low C3)
- Postinfectious, SLE, cryoglobulinemia, subacute bacterial endocarditis (SBE), hepatitis B/C, syphilis, schistosomiasis, tumor
- Low C3 levels (complement consumed by circulating immune complexes)
- Cryoglobulinemia: also low C4 + high rheumatoid factor (hepatitis C or paraproteinemia)
- IgA vasculitis (HSP): palpable purpura, GI bleeding, arthralgias, ± pulmonary hemorrhage; C3 in mesangium, serum C3 normal
4. Lupus Nephritis
"Full-house" immunofluorescence (all immunoglobulins + C3 + C1q). WHO/ISN/RPS class III-IV (diffuse proliferative) is the most aggressive and requires immunosuppression.
5. Membranoproliferative GN (MPGN)
"Double contouring" of the GBM on PAS stain. Immune complex-mediated subendothelial deposits. Progressive course.
6. C3 Glomerulopathy (C3GN / Dense Deposit Disease)
Caused by dysregulation of the alternative complement pathway (e.g., C3 nephritic factor, factor H deficiency). Dominant C3 staining with minimal immunoglobulin.
Relative Frequency of RPGN by Age
From Brenner & Rector's The Kidney, pauci-immune crescentic GN (ANCA-associated) is the most common type overall, particularly in older adults. Anti-GBM disease is more common in young adults, and immune complex GN predominates in children and younger adults.
Treatment Principles
| Type | Treatment |
|---|---|
| PIGN | Supportive; antibiotics only if ongoing infection; resolves spontaneously in most cases |
| IgA nephropathy | ACE-I/ARBs (all patients); corticosteroids ± fish oils; targeted complement therapy for progressive disease |
| Anti-GBM disease | Plasmapheresis + cyclophosphamide + corticosteroids (urgent) |
| ANCA-associated RPGN | High-dose corticosteroids + cyclophosphamide or rituximab (induction); maintenance with lower-dose steroids + azathioprine or rituximab |
| Immune complex RPGN | IV methylprednisolone (1-3 g) → oral corticosteroids + cyclophosphamide; less responsive than anti-GBM/ANCA types |
| Lupus nephritis | Corticosteroids + mycophenolate mofetil (class III/IV); belimumab/voclosporin as add-on |
Key note from Brenner & Rector: immune complex crescentic GN is less responsive to aggressive immunosuppressive therapy than anti-GBM or ANCA crescentic GN.
Complement Levels as a Diagnostic Clue
| Condition | C3 | C4 |
|---|---|---|
| PIGN | Low | Low initially, then normal |
| SLE nephritis | Low | Low |
| Cryoglobulinemia | Low | Low |
| IgA nephropathy | Normal | Normal |
| Anti-GBM / ANCA | Normal | Normal |
Key Summary
- GN spans a spectrum from self-limited (PIGN in children) to rapidly progressive (crescentic GN) to chronic (IgAN, MPGN)
- The biopsy with light microscopy, immunofluorescence, and electron microscopy remains essential for definitive diagnosis
- Complement levels (C3, C4) and serologies (ANCA, anti-GBM, ANA, cryoglobulins, ASO) guide initial categorization before biopsy
- Early diagnosis and treatment in crescentic GN can reverse potentially fatal renal failure
Sources: Harrison's Principles of Internal Medicine 22E, p. 2397-2399; Robbins & Kumar Basic Pathology; Henry's Clinical Diagnosis and Management, p. 1249; Brenner and Rector's The Kidney
This is a shared conversation. Sign in to Orris to start your own chat.