Gitelman syndrome
Reading File
Finding Sources
Finding Sources
Searching PubMed
Gitelman syndrome
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Searching Images
Gitelman syndrome distal convoluted tubule NCC cotransporter mechanism
I have all the information needed. Let me compile a comprehensive answer.
Gitelman Syndrome
Gitelman syndrome (GS; OMIM #263800) is the most common inherited renal tubular disorder, with a prevalence of ~25 per million (heterozygous carrier rate ~1% of the general population). It is an autosomal recessive salt-losing tubulopathy affecting the distal convoluted tubule (DCT).
Genetics and Molecular Basis
| Gene | Protein | Location | Notes |
|---|---|---|---|
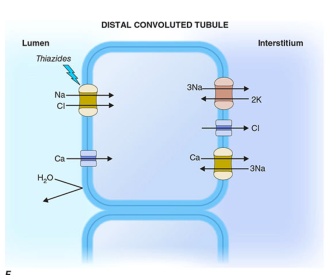
| SLC12A3 | Thiazide-sensitive Na⁺-Cl⁻ cotransporter (NCC/NCCT) | Apical membrane, DCT | Responsible for the vast majority of cases |
| CLCNKB | Basolateral chloride channel CLC-Kb | Basolateral membrane, DCT/TAL | Rare; phenotype overlaps with Bartter type 3 |
Loss-of-function mutations in SLC12A3 abolish NCC activity. Many mutations cause a defect in cellular trafficking (the mutant protein is retained in the endoplasmic reticulum and never reaches the apical membrane). Commercial genetic panels now test the full complement of relevant genes rather than single genes.
Pathophysiology
The mechanisms mirror those of chronic thiazide diuretic use:
- Loss of NCC function → Na⁺ and Cl⁻ wasting from the DCT → volume depletion
- Volume depletion → activates the renin–angiotensin–aldosterone system (RAAS)
- Increased aldosterone → upregulates ENaC in the cortical collecting tubule (CNT) → increased Na⁺ reabsorption, offset by K⁺ and H⁺ secretion → hypokalemia + metabolic alkalosis
- Hypomagnesemia: NCC deficiency leads to DCT cell apoptosis and downregulation of the epithelial Mg²⁺ channel TRPM6 in the DCT → renal Mg²⁺ wasting
- Hypocalciuria: Volume contraction enhances proximal tubular Ca²⁺ reabsorption (passive, secondary to increased Na⁺ reabsorption); the late DCT remains morphologically intact with preserved TRPV5 expression
Clinical Features
GS presents in childhood or adolescence (contrast with Bartter syndrome, which presents neonatally). Many patients are discovered incidentally on routine electrolyte testing.
Symptoms (variable severity):
- Muscle weakness, cramps, fatigue (most common)
- Salt craving (nearly universal; sometimes extreme)
- Nocturia
- Polyuria and polydipsia (rare — distinguishes GS from Bartter)
- Paresthesias, tetany (exceptional)
- QT interval prolongation (increased arrhythmia risk; rare events of presyncope/ventricular tachycardia reported)
- Chondrocalcinosis (calcium pyrophosphate dihydrate deposition in joint cartilage — a distinctive late complication, linked to hypomagnesemia)
- Sclerochoroidal calcifications (ocular choroidal calcification)
- Women: symptom exacerbation with menses; complicated pregnancies possible
Blood pressure: Normal to low (no hypertension)
Laboratory Findings
| Parameter | Finding |
|---|---|
| Serum K⁺ | Low (hypokalemia) |
| Serum Mg²⁺ | Low (hypomagnesemia) |
| Serum HCO₃⁻/pH | High (metabolic alkalosis) |
| Serum Cl⁻ | Low (hypochloremia) |
| Serum Ca²⁺ | Normal |
| Urine Ca²⁺ | Low (hypocalciuria) — key distinguishing feature |
| Urine Cl⁻, K⁺, Mg²⁺ | Elevated (renal wasting) |
| Renin/Aldosterone | Elevated (secondary hyperreninemic hyperaldosteronism) |
| Urinary prostaglandin E₂ | Normal (unlike Bartter syndrome) |
| Blood pressure | Normal to low |
Distinguishing Gitelman from Bartter Syndrome
| Feature | Gitelman | Bartter |
|---|---|---|
| Defect site | DCT (NCC) | TAL (NKCC2, ROMK, CLC-Kb, Barttin) |
| Age of onset | Childhood/adolescence | Neonatal |
| Urinary calcium | Low | Elevated |
| Serum magnesium | Low | Normal |
| Nephrocalcinosis | Absent | Present |
| Polyhydramnios | Absent | Common |
| Polyuria/polydipsia | Rare | Marked |
| Urinary PGE₂ | Normal | Elevated |
| Response to NSAIDs | Poor | Good |
Differential Diagnosis
- Bartter syndrome (especially type 3, which can overlap)
- EAST/SeSAME syndrome (KCNJ10 mutation: hypokalemic alkalosis + epilepsy, ataxia, sensorineural deafness, mental retardation)
- Thiazide diuretic abuse (identical biochemical picture — check urine diuretic screen)
- Laxative abuse, chronic vomiting (urine Cl⁻ helps distinguish: low in vomiting, elevated in GS)
- Sjögren syndrome (acquired GS phenotype: hypokalemia + hypomagnesemia + hypocalciuria with normal NCC coding sequence)
- HNF1B-related disease
- Cisplatin nephrotoxicity (can cause an acquired GS-like picture)
Treatment
Targets: Serum K⁺ ≥ 3.0 mEq/L; Serum Mg²⁺ ≥ 0.6 mmol/L (1.46 mg/dL)
- Dietary salt liberalization — counteracts volume depletion
- Oral magnesium supplementation (lifelong): Organic magnesium salts (e.g., magnesium citrate, glycinate) preferred over MgCl₂, Mg(OH)₂, or MgO (better tolerated, less diarrhea)
- Potassium supplementation: Oral KCl or potassium-rich diet
- Amiloride — often preferred over spironolactone; blocks ENaC, reduces kaliuresis; doses up to 10 mg twice daily; can be combined with spironolactone or eplerenone
- Spironolactone / eplerenone — adjunct to amiloride for refractory hypokalemia
- RAAS inhibitors (ACE inhibitors) — in selected patients where hypotension is not a concern
- NSAIDs — generally not useful (prostaglandin levels are normal, unlike Bartter syndrome)
- Dietary caution: Identify and limit excessive salt-loading foods — salt loading increases distal NaCl delivery, amplifying K⁺ secretion by the CCT
Bone Effects
An interesting paradox: homozygous GS patients show higher bone mineral density than unaffected wild-type family members, while heterozygotes have intermediate values. This mirrors the bone-protective effect of thiazide diuretics. The hypocalciuria and enhanced proximal Ca²⁺ reabsorption are presumed responsible.
Acquired (Secondary) Gitelman-Like Syndromes
Conditions that mimic GS biochemically without NCC mutations:
- Sjögren syndrome with tubulointerstitial nephritis
- Cisplatin chemotherapy
- Autoimmune tubulointerstitial nephritis
Sources: Brenner and Rector's The Kidney (2-Volume Set); National Kidney Foundation Primer on Kidney Diseases, 8e; Comprehensive Clinical Nephrology, 7th Edition; Harrison's Principles of Internal Medicine, 22e
This is a shared conversation. Sign in to Orris to start your own chat.