Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have everything needed for a comprehensive answer. Let me compile it.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is the clinical manifestation of severe acute lung injury (ALI) — defined by the Berlin criteria as bilateral infiltrates, a PaO₂/FiO₂ ratio <300, onset within 7 days of a known insult, and absence of a purely cardiogenic cause. Its pathological substrate is diffuse alveolar damage (DAD). The mechanism unfolds in three overlapping phases.
1. Triggering Insults
Over 50% of ARDS cases stem from four conditions: sepsis, diffuse pulmonary infections (viral, bacterial, fungal), gastric aspiration, and major trauma. Other triggers include pancreatitis, massive transfusion (TRALI), burns, inhalation injury, and drug toxicity. Insults can act directly on the lung (pneumonia, aspiration) or indirectly via the systemic circulation (sepsis, trauma releasing circulating mediators).
2. Initiation: Alveolar-Capillary Interface Injury
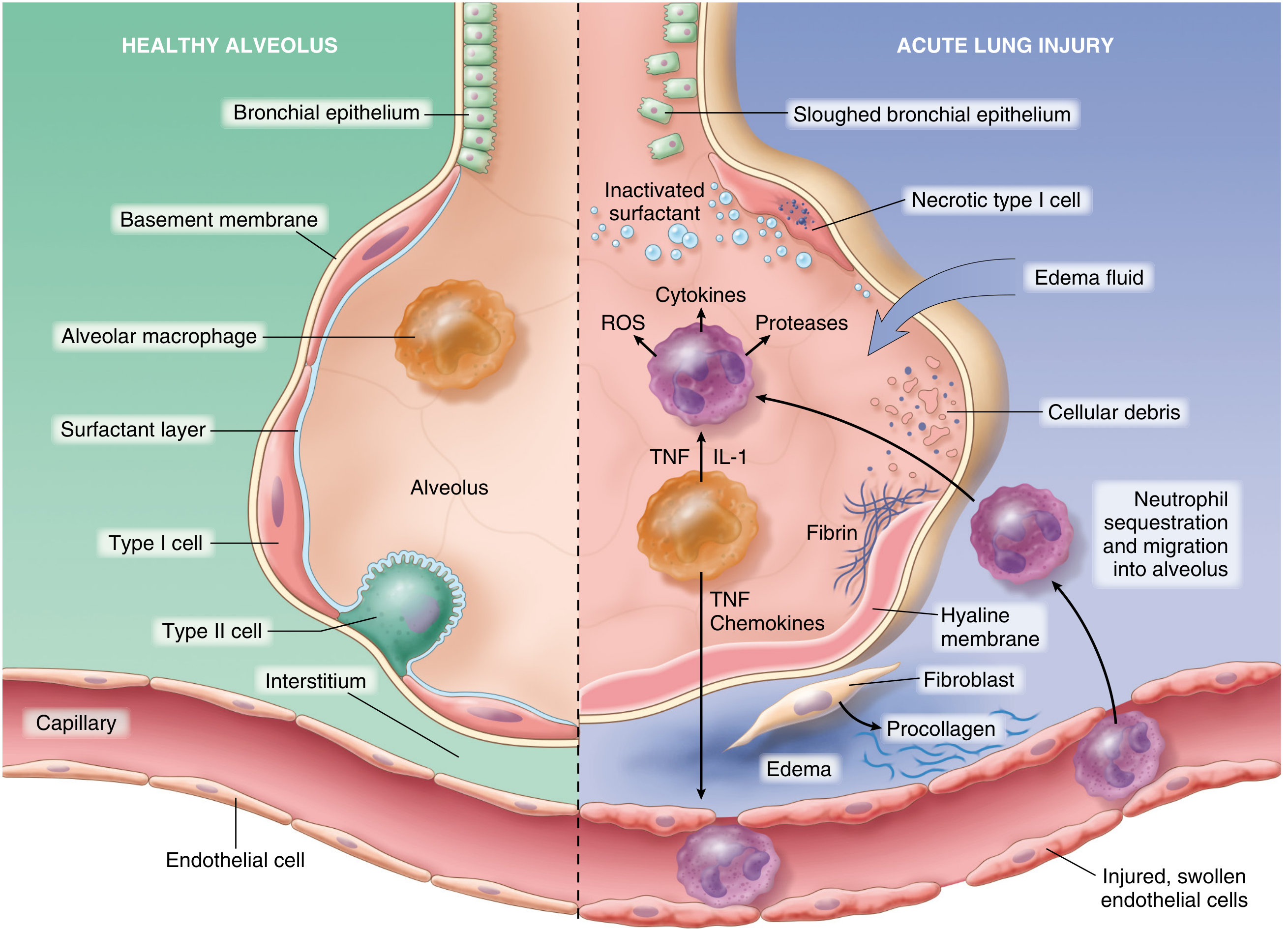
ALI/ARDS is initiated by concurrent injury to type I pneumocytes and pulmonary endothelium, setting off a self-amplifying inflammatory loop:
Endothelial Activation
- Pneumocyte injury is sensed by resident alveolar macrophages, which release TNF-α and IL-1 that act on adjacent endothelium.
- In sepsis or severe trauma, circulating inflammatory mediators activate pulmonary endothelium directly.
- Activated endothelium upregulates adhesion molecules (E-selectin, ICAM-1), procoagulant proteins, and chemokines (IL-8).
Neutrophil Sequestration and Degranulation
- Neutrophils adhere to activated endothelium and extravasate into the interstitium and alveoli.
- They degranulate, releasing proteases (elastase, collagenase), reactive oxygen species (ROS), and further cytokines.
- Neutrophil extracellular traps (NETs) are released, causing direct structural lung damage.
- This creates a positive feedback loop: more inflammation → more endothelial damage → more neutrophil recruitment.
3. Loss of Alveolar-Capillary Barrier Integrity
- Endothelial injury makes pulmonary capillaries leaky → protein-rich fluid floods the interstitium and alveolar spaces (noncardiogenic pulmonary edema).
- Necrosis of type II pneumocytes disrupts surfactant production and function → alveolar collapse and reduced compliance.
- Loss of type I pneumocytes (which cover ~95% of the alveolar surface) eliminates the gas exchange epithelium.
4. Acute (Exudative) Phase — Days 1–6: Hyaline Membrane Formation
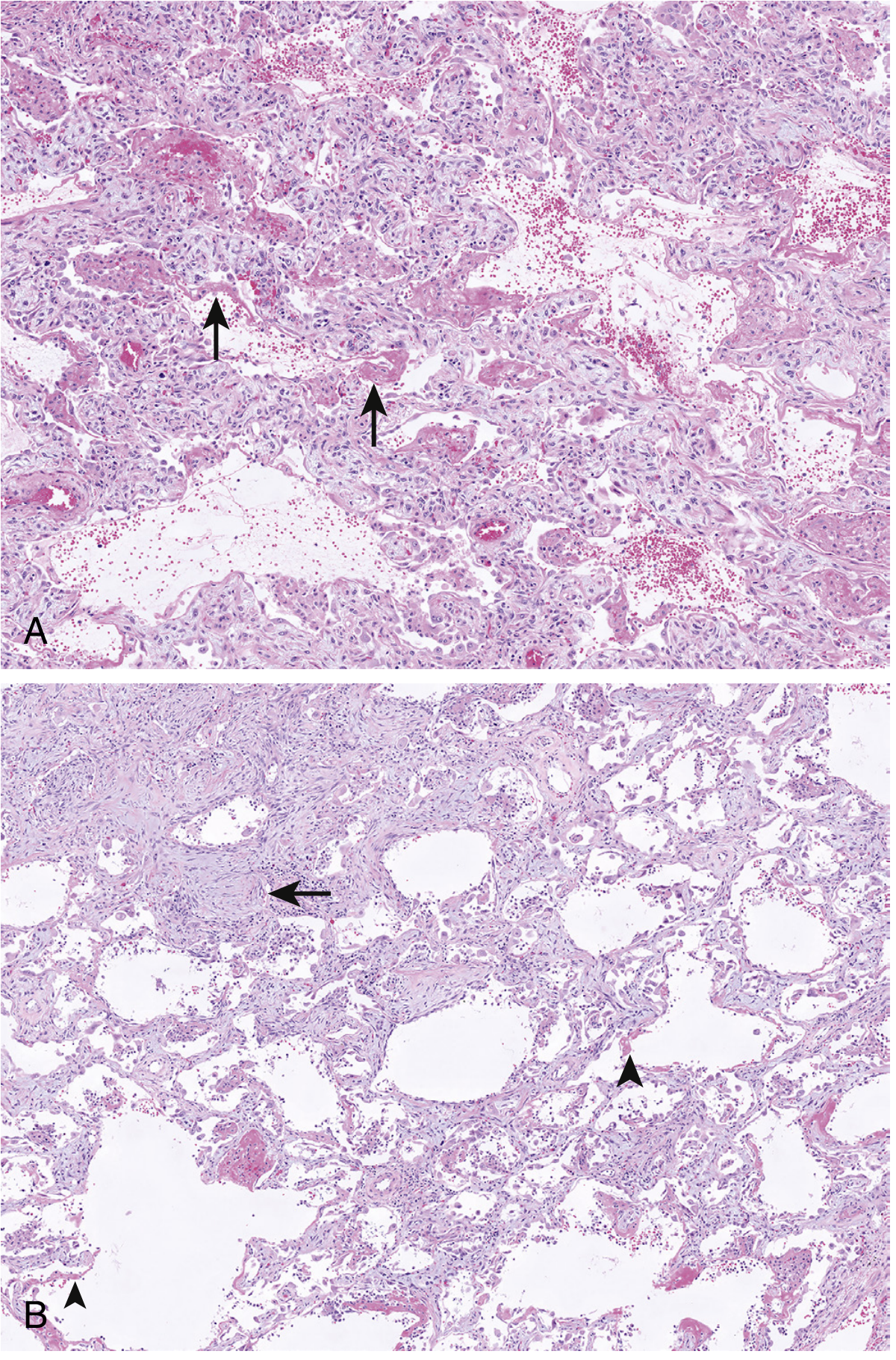
- Protein-rich exudate + fibrin + necrotic cell debris organizes into hyaline membranes lining the alveolar walls — the histopathologic hallmark of DAD.
- Alveolar septa show interstitial edema with scattered inflammatory cells in septal capillaries.
- The alveolar space contains proteinaceous exudate, fibrin, macrophages, and scattered neutrophils.
5. Organizing (Proliferative) Phase — Days 7–21
- Fibroblasts proliferate within thickened alveolar septa (fibroblast-rich granulation tissue), driven by TGF-β and PDGF released from macrophages clearing debris.
- Type II pneumocyte hyperplasia occurs as these cells attempt re-epithelialization of the damaged alveolar surface.
- Intraalveolar exudates are replaced by rounded plugs of granulation tissue (resembling organizing pneumonia).
6. Resolution or Fibrosis
- If the trigger abates and inflammation resolves, macrophages clear debris, type II pneumocytes differentiate into type I cells, and capillary endothelium regenerates — sometimes with near-complete recovery.
- In other patients, progressive collagen deposition leads to irreversible fibrosis and impaired cardiopulmonary function.
7. Physiological Consequences
| Mechanism | Consequence |
|---|---|
| Alveolar flooding + collapse | ↓ Functional residual capacity (FRC) |
| Surfactant loss | ↓ Compliance, alveolar instability |
| V/Q mismatch (flooded alveoli perfused but not ventilated) | Refractory hypoxemia |
| Increased dead space (damaged capillaries) | Hypercapnia in severe disease |
| Heterogeneous disease distribution (gravity-dependent consolidation) | "Baby lung" phenomenon — only ~30% of lung available for ventilation |
The V/Q mismatch is the principal cause of hypoxemia: poorly aerated regions continue to receive blood flow, shunting deoxygenated blood into the systemic circulation. Because this is an intrapulmonary shunt (not a diffusion limitation), it responds poorly to supplemental oxygen alone, which is the hallmark of ARDS hypoxemia.
8. Coagulation Dysfunction
- Endothelial injury triggers local microvascular thrombosis via tissue factor expression and fibrin deposition within alveolar capillaries.
- This further reduces perfusion to affected regions and worsens gas exchange.
- Platelet-neutrophil complexes (via P-selectin/PSGL-1) amplify ROS and NET release.
Summary Cascade
Sepsis / Aspiration / Trauma / Infection
↓
Alveolar macrophage activation → TNF-α, IL-1, IL-8
↓
Endothelial activation → ↑Adhesion molecules, procoagulants
↓
Neutrophil sequestration → ROS, proteases, NETs
↓
Alveolar-capillary barrier breakdown
↓
Protein-rich edema + surfactant failure + type I pneumocyte necrosis
↓
Hyaline membrane formation (DAD)
↓
V/Q mismatch → Refractory hypoxemia (ARDS)
↓
Resolution ←→ Fibroproliferation
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 15 (ALI/ARDS Pathogenesis)
- Murray & Nadel's Textbook of Respiratory Medicine, Chapters 22 & 94 (Diffuse Alveolar Damage)
- Sabiston Textbook of Surgery, Chapter 11 (ARDS in Critical Care)
This is a shared conversation. Sign in to Orris to start your own chat.