urticaria
Reading File
Finding Sources
Searching PubMed
urticaria management guidelines
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
I now have comprehensive content from multiple authoritative textbooks. Let me compile a thorough response.
Urticaria (Hives)
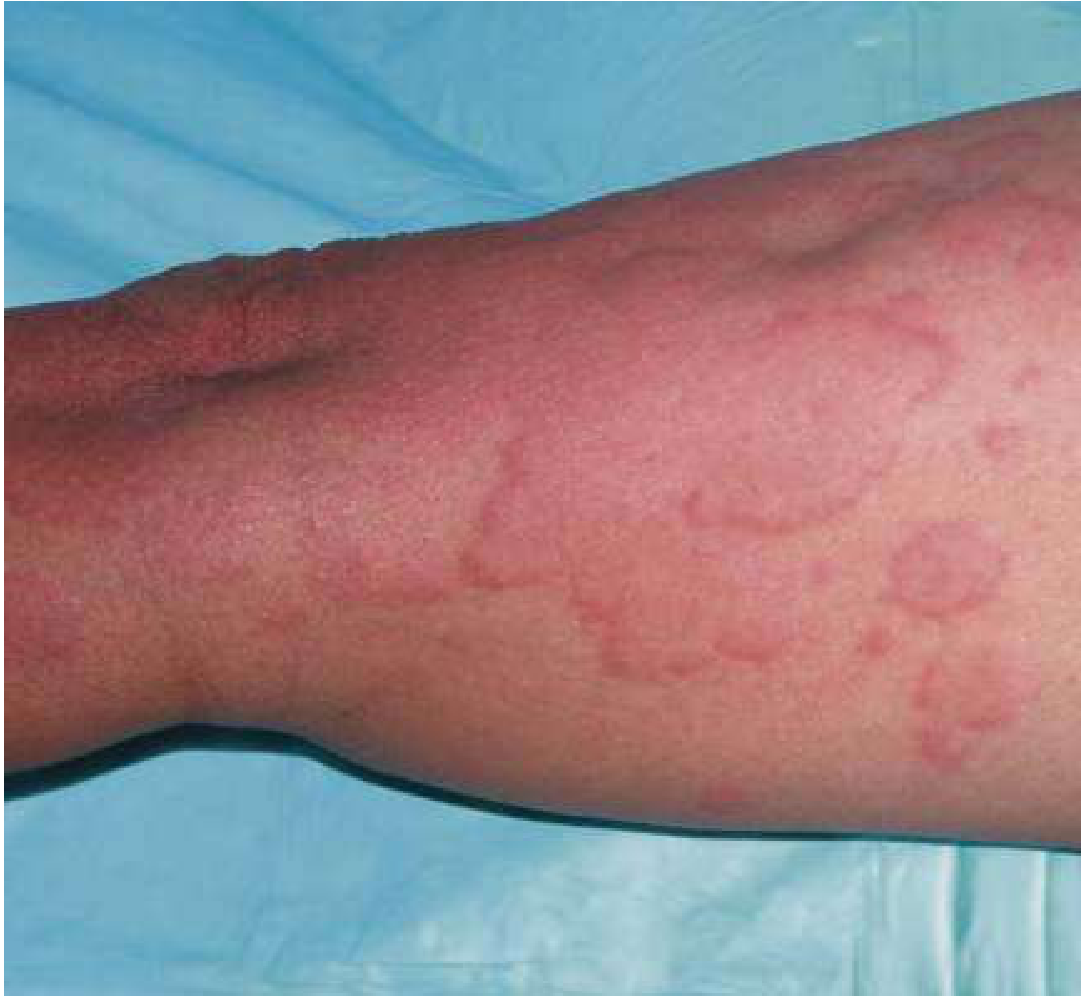
Urticaria: erythematous, raised, blanching wheals — Rosen's Emergency Medicine
Definition & Epidemiology
Urticaria (hives) is one of the most common cutaneous reaction patterns. Lesions are pink to light red, raised, blanching, pruritic wheals with pale centers and red borders. A defining feature is that individual hives persist <24 hours, though new lesions continually develop.
- Lifetime prevalence: ~15–20% of the population
- Acute urticaria: episodes lasting <6 weeks; common in children, both sexes
- Chronic urticaria: recurrence ≥6 weeks; peaks in the 3rd–4th decade, more common in women 40s–50s; 50% persist ≥5 years
— Goldman-Cecil Medicine, p. 4303; Rosen's Emergency Medicine, p. 2413
Pathobiology
The final common pathway is mast cell degranulation releasing histamine, slow-reacting substance of anaphylaxis (leukotrienes LTC4, LTD4), bradykinin, and kallikrein → localized dermal edema from transvascular fluid extravasation.
Immunologic mechanisms:
- IgE-mediated (type I hypersensitivity) — allergen crosslinks IgE on mast cells
- Functional IgE autoantibodies — found in chronic urticaria, directly release histamine from mast cells and basophils
- Immune complex-mediated (type III)
- Complement-kinin dependent
Non-immunologic mechanisms:
- Direct mast cell degranulators — opiates, radiocontrast media, strawberries, lobster
- Prostaglandin pathway — aspirin, NSAIDs
- Physical stimuli
Basophils are recruited into wheals and sustain the histamine response. Eosinophils contribute via LTC4/LTD4.
— Goldman-Cecil Medicine, p. 4303–4304
Classification & Common Causes
Spontaneous Urticaria
| Type | Cause |
|---|---|
| Acute (<6 wk) | Drugs (penicillin, sulfa, NSAIDs, opiates), foods (shellfish, nuts, eggs, berries), infections, latex, blood products |
| Chronic spontaneous | Often idiopathic; autoimmune (IgG anti-FcεRI or anti-IgE autoantibodies); occult infections (H. pylori, sinusitis, dental abscesses, viral hepatitis) |
Physical Urticarias
| Type | Trigger | Key Features |
|---|---|---|
| Dermographism | Firm skin stroking | Wheal within 30 min; most common form |
| Pressure urticaria | Sustained pressure | Onset delayed 4–8 h |
| Cold urticaria | Ice/cold water | Ice cube test positive; potentially life-threatening on immersion |
| Cholinergic urticaria | Exercise, heat, emotion | Small 1–3 mm wheals with large erythematous flares |
| Solar urticaria | Sun-exposed areas | Clears when light removed |
| Aquagenic urticaria | Water contact | Rare |
| Vibratory angioedema | Vibration | Swelling within minutes, lasts ~30 min |
Infections as Triggers
Rhinovirus, rotavirus, hepatitis B/C, EBV mononucleosis, coxsackievirus, Candida, dermatophytes, streptococcal pharyngitis (especially in children)
Systemic Disease Associations
SLE, rheumatoid arthritis, rheumatic fever (erythema marginatum), hyperthyroidism, lymphoma/Hodgkin disease, mastocytosis, C1 esterase inhibitor deficiency
— Rosen's Emergency Medicine, p. 2413–2414; Goldman-Cecil Medicine, p. 4304
Clinical Manifestations
- Edematous plaques, pale center, red/pink border ("wheal and flare")
- Individual lesions resolve within <24 hours (distinguishes from vasculitis)
- May coalesce into giant plaques or annular rings
- Angioedema: deeper swelling of subcutaneous tissue or mucous membranes; accompanies urticaria in many cases
- Anaphylaxis with laryngeal edema = life-threatening emergency
- Pruritus may precede visible lesions by minutes to hours
Diagnosis
History is critical: timing (minutes to hours after exposure), medications (including OTC, supplements), foods, occupation, prior atopy, duration.
Key diagnostic points:
- Lesions lasting >24 hours → suspect urticarial vasculitis (skin biopsy required; leukocytoclastic vasculitis on pathology)
- Darier sign (stroking a lesion induces urticaria) → suspect mastocytosis (urticaria pigmentosa); elevated serum tryptase/histamine
- For chronic urticaria work-up: CBC, ESR, LFTs, TFTs, ANA, complement levels, Helicobacter serology, allergy testing if history is unrevealing
Differential diagnosis: Drug eruption, viral exanthem, erythema multiforme, erythema marginatum, juvenile rheumatoid arthritis rash, mastocytosis
— Goldman-Cecil Medicine, p. 4304; Rosen's Emergency Medicine, p. 2414
Treatment
Step 1 — Remove/Avoid the Trigger
Avoid aspirin, NSAIDs (non-immunologically worsen urticaria), offending foods, contactants.
Step 2 — Antihistamines (First-Line)
- Second-generation H1 antihistamines (preferred — non-sedating): cetirizine, fexofenadine, loratadine
- First-generation H1 (diphenhydramine, hydroxyzine): use for acute/severe or where sedation is acceptable
- Adding an H2 blocker (ranitidine/famotidine) may provide additive benefit
Step 3 — Corticosteroids
- Short courses (prednisone up to 1 mg/kg) for moderate–severe acute urticaria
- 14–21 day taper for recurrent urticaria
- Chronic systemic corticosteroids are not recommended due to long-term toxicity
Step 4 — Urticaria with Anaphylaxis/Wheezing
- Epinephrine SC/IM immediately
- IV corticosteroids
- Oxygen, airway management
Step 5 — Refractory Chronic Spontaneous Urticaria
- Omalizumab (anti-IgE monoclonal antibody) 300 mg SC monthly — effective in 65–70% of antihistamine-refractory patients
- Cyclosporine 3 mg/kg for 8–16 weeks — similar efficacy
- Physical urticarias: pre-treatment with antihistamines 30–60 min before known trigger (e.g., cold urticaria); phototherapy to induce tolerance (solar urticaria)
— Goldman-Cecil Medicine, p. 4304; Rosen's Emergency Medicine, p. 2414
Angioedema — Key Distinction
| Feature | Urticaria | Hereditary Angioedema (C1-INH deficiency) |
|---|---|---|
| Mechanism | Mast cell/histamine | Bradykinin-mediated |
| Pruritus | Yes | Minimal |
| Response to antihistamines | Yes | Poor |
| Treatment | Antihistamines, steroids | C1-INH concentrate, icatibant, tranexamic acid |
Recent Evidence
Recent systematic reviews (2024–2025) are updating practice:
- Treatment of acute urticaria (2024) — systematic review confirming evidence base for H1-antihistamines as first-line (PMID 38420865)
- Clinical practice guideline for H1-antihistamine–resistant chronic spontaneous urticaria (2025) — covers step-up therapies including omalizumab and beyond (PMID 41182242)
- A 2025 systematic review (PMID 40742623) identifies poor adherence to antihistamine therapy as a major obstacle in chronic spontaneous urticaria management
Sources: Goldman-Cecil Medicine International Edition (Chapter 407) | Rosen's Emergency Medicine (Chapter 107) | Fitzpatrick's Dermatology Vol. 1 | Textbook of Family Medicine 9e
What are the mechanisms of action of omalizumab and cyclosporine in refractory urticaria?
Finding Sources
Finding Sources
Searching PubMed
omalizumab cyclosporine chronic spontaneous urticaria mechanism
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading Article
Reading File
Reading File
Loading Image
I now have everything needed for a comprehensive, well-illustrated answer. The Fitzpatrick's diagram is excellent and perfectly illustrates the omalizumab mechanism. Let me compile the full response.
Mechanisms of Action in Refractory Chronic Urticaria
Omalizumab (Anti-IgE Monoclonal Antibody)
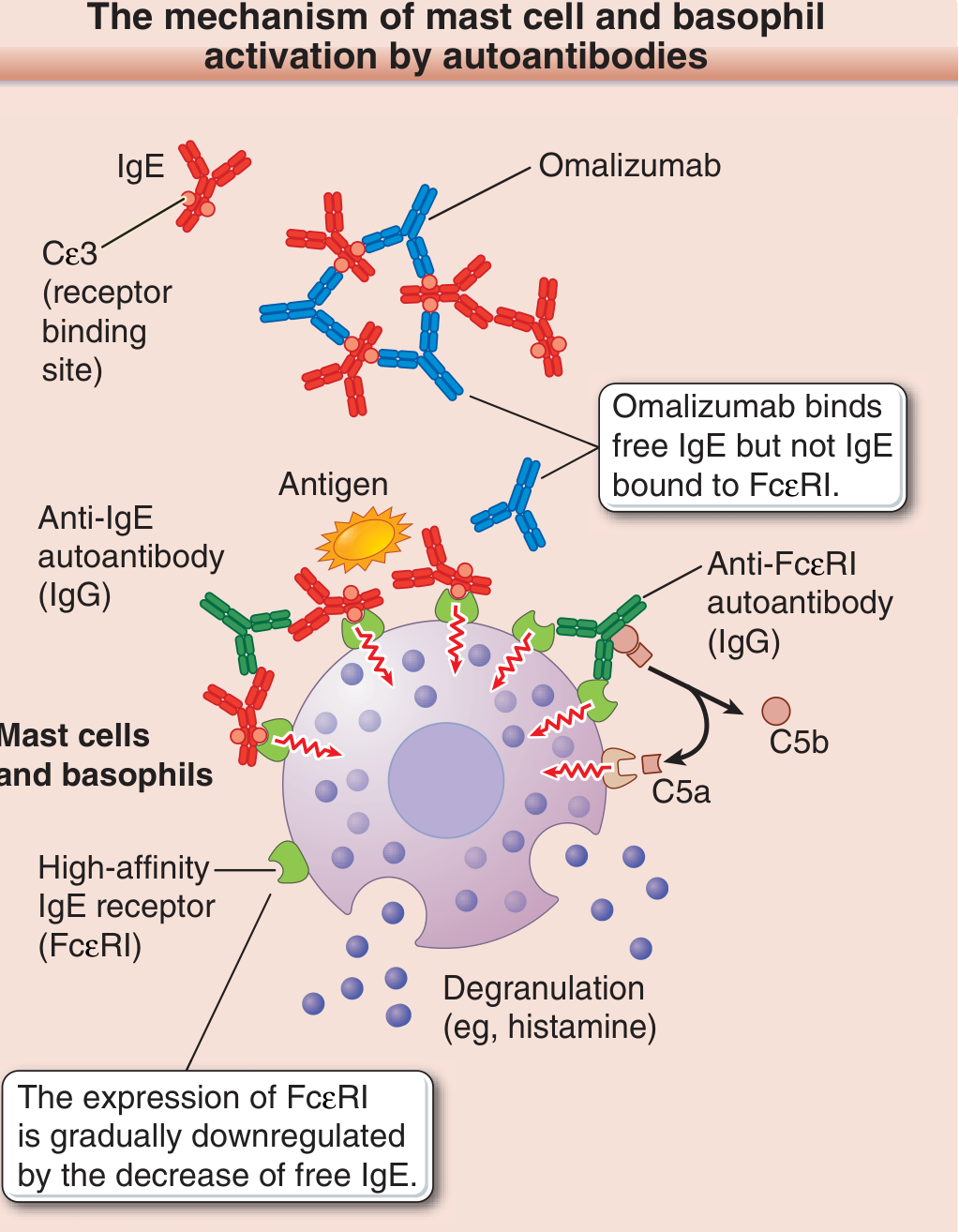
Background: Why IgE is the target
In chronic spontaneous urticaria (CSU), mast cells and basophils are activated through multiple IgE-dependent mechanisms:
- Exogenous antigen crosslinks antigen-specific IgE bound to FcεRI → mast cell degranulation
- IgG autoantibodies against IgE — crosslink IgE molecules already bound to FcεRI, triggering degranulation
- IgG autoantibodies against FcεRI — bind directly to and crosslink the receptor; their histamine-releasing activity is partially complement-dependent (C5a is generated, which is itself a direct mast cell activator)
Approximately one-third to one-half of CSU patients carry these functional IgG autoantibodies. The rapid effect of omalizumab even on physical urticarias suggests additional endogenous IgE-bound autoantigens drive pathogenesis beyond what is currently characterized.
— Fitzpatrick's Dermatology, Vol. 1, p. 720–721
Omalizumab's mechanism (multi-step)
Step 1 — Binding of free IgE:
Omalizumab is a humanized monoclonal antibody that binds the Cε3 domain of free (circulating) IgE — the same region that normally docks to FcεRI. Crucially, it cannot bind IgE that is already attached to FcεRI on mast cells or basophils (which would risk triggering degranulation). This selectivity makes it safe.
Step 2 — Reduction of free IgE:
By sequestering free IgE into omalizumab-IgE immune complexes, circulating free IgE levels fall dramatically (>95%).
Step 3 — FcεRI downregulation:
FcεRI expression on mast cells and basophils is regulated by ambient free IgE levels — the receptor is stabilized by IgE occupancy. As free IgE falls, FcεRI surface density on mast cells and basophils decreases (receptor downregulation). Fewer receptors mean less capacity for crosslinking-driven activation, regardless of the triggering mechanism.
Step 4 — Reduced autoantibody-mediated activation:
With fewer FcεRI receptors and less surface-bound IgE available, the IgG autoantibodies against IgE or FcεRI (the autoimmune drivers of CSU) lose their targets → reduced mast cell/basophil degranulation.
Clinical implication: Patients with a positive autologous serum skin test (ASST) — indicating autoantibody-driven (type IIb autoimmune) disease — respond to omalizumab more slowly than ASST-negative patients, consistent with the time needed for FcεRI downregulation. Efficacy: ~65–70% of antihistamine-refractory patients respond.
— Fitzpatrick's Dermatology, p. 721; Goldman-Cecil Medicine, p. 4304
Cyclosporine (Calcineurin Inhibitor)
Molecular mechanism
Step 1 — Binding to cyclophilin:
Cyclosporine, a cyclic fungal peptide, enters cells and binds with high affinity to cyclophilin (an intracellular immunophilin).
Step 2 — Calcineurin inhibition:
The cyclosporine-cyclophilin complex binds to and inhibits calcineurin (a Ca²⁺/calmodulin-activated serine/threonine phosphatase). Calcineurin's normal job is to dephosphorylate the cytosolic form of NFAT (nuclear factor of activated T cells), which allows NFAT to translocate into the nucleus.
Step 3 — Blockade of cytokine gene transcription:
With calcineurin blocked, cytoplasmic NFAT remains phosphorylated and cannot enter the nucleus → transcription of IL-2 and other cytokine genes is suppressed → T-cell activation and proliferation are blocked.
— Cellular and Molecular Immunology (Abbas), p. 1145; Goodman & Gilman's, p. 791; Lippincott Pharmacology, p. 3426
How this applies to urticaria
Cyclosporine's benefit in CSU comes from multiple complementary effects beyond T-cell suppression:
| Target | Effect |
|---|---|
| T helper cells | ↓ IL-2, IL-4, IL-5 → reduced B cell help → fewer autoantibodies produced |
| Mast cells | Direct inhibition of mast cell degranulation (calcineurin is also expressed in mast cells; NFAT drives mast cell cytokine production) |
| Basophils | Reduced IgG autoantibody-driven basophil activation |
| IgG autoantibody production | Reduced anti-FcεRI and anti-IgE autoantibody titers over time |
The rationale is therefore upstream immunomodulation: cyclosporine suppresses the T-cell-driven autoimmune process that generates the pathogenic IgG autoantibodies, while also directly dampening effector cell activity.
Comparison at a Glance
| Feature | Omalizumab | Cyclosporine |
|---|---|---|
| Class | Humanized monoclonal anti-IgE Ab | Calcineurin inhibitor (immunosuppressant) |
| Primary target | Free circulating IgE (Cε3 domain) | Cyclophilin → calcineurin → NFAT |
| Downstream effect | ↓ FcεRI surface expression; ↓ mast cell/basophil sensitivity | ↓ IL-2 → ↓ T-cell proliferation; ↓ autoantibody production; direct mast cell suppression |
| Onset | Rapid (within days–weeks); slower in ASST+ patients | Weeks (immunosuppression must accumulate) |
| Guideline position | Preferred 3rd-line (EAACI) | Alternative 3rd-line |
| Dose in CSU | 300 mg SC monthly | 3 mg/kg/day for 8–16 weeks |
| Key AEs | Injection site reactions, rare anaphylaxis | Nephrotoxicity, hypertension, drug interactions |
| Efficacy | 65–70% of antihistamine-refractory patients | 65–70% (similar) |
Emerging Perspective (2024)
A 2024 Lancet review (Zuberbier et al., PMID 39004090) identifies the limitation of this linear step-up approach and proposes endotype-based personalization: patients with type I autoimmunity (IgE against autoantigens) may respond better to omalizumab, while those with type IIb autoimmunity (IgG anti-FcεRI/IgE) may need immunosuppression (cyclosporine, or emerging BTK inhibitors) to address the upstream autoantibody production.
Sources: Fitzpatrick's Dermatology Vol. 1, p. 720–721 | Goldman-Cecil Medicine, p. 4304 | Cellular and Molecular Immunology (Abbas), p. 1145 | Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 791
This is a shared conversation. Sign in to Orris to start your own chat.