Types of rental tubular acidosis, their presentation and management in wards
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Searching PubMed
"renal tubular acidosis" AND management
I have all the information needed for a comprehensive answer. Let me compile the full response now.
Renal Tubular Acidosis (RTA): Types, Presentation, and Ward Management
RTA is defined as a hyperchloremic (normal anion gap) metabolic acidosis caused by defects in renal tubular handling of bicarbonate or hydrogen ion, in the setting of a normal or near-normal GFR. There are three clinically recognized types: Type 1 (distal), Type 2 (proximal), and Type 4 (hyperkalemic distal). A Type 3 (mixed) is sometimes listed but is rarely used clinically.
Pathophysiology Overview
The kidney maintains acid-base balance through two main mechanisms:
- Proximal tubule: reabsorbs ~85-90% of filtered bicarbonate (via Na+/H+ exchanger and carbonic anhydrase)
- Distal nephron (alpha-intercalated cells): secretes net H+ into urine via H+-ATPase and H+/K+-ATPase, and generates new bicarbonate via AE1 at the basolateral membrane
Failure of either mechanism causes metabolic acidosis with a normal anion gap.
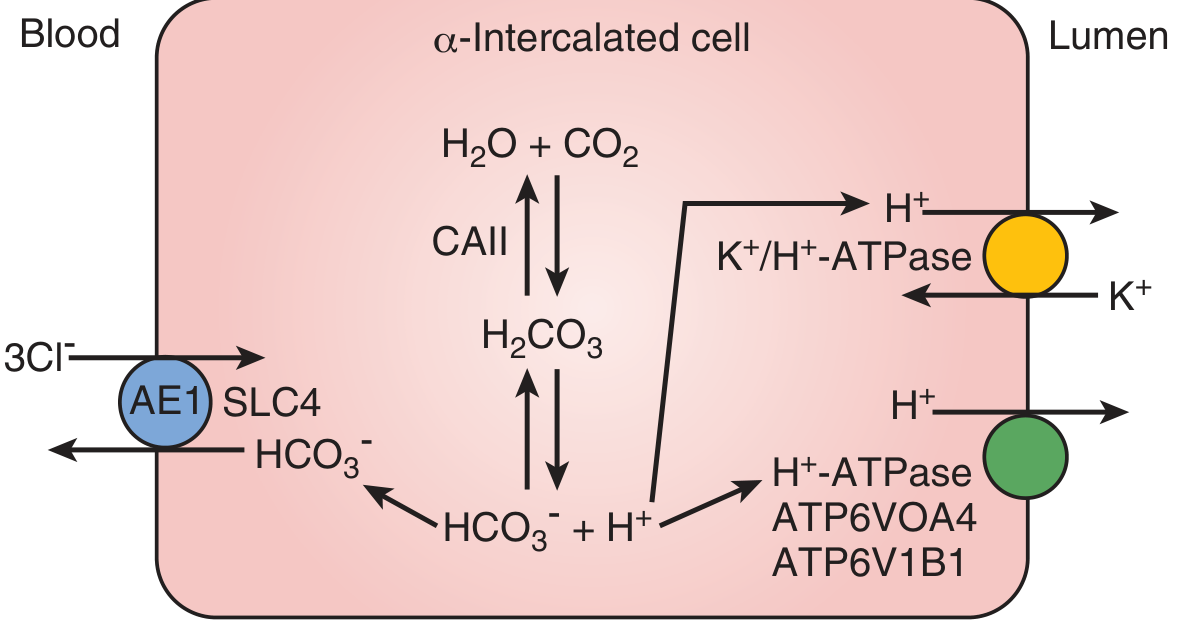
Type 1 - Distal RTA (dRTA)
Pathophysiology
Dysfunction of alpha-intercalated cells in the collecting duct. The H+-ATPase (or its subunits ATP6V1B1, ATP6V0A4) or the basolateral AE1 (SLC4A1) Cl-/HCO3- exchanger are defective. The kidney cannot acidify urine below pH 5.5 regardless of the degree of systemic acidosis. Loss of electrogenic H+ secretion promotes K+ secretion to maintain electronegativity, driving hypokalemia. - Campbell Walsh Wein Urology, p. 2688
Causes
| Inherited | Acquired |
|---|---|
| AD: SLC4A1 mutation | Sjogren syndrome (most common adult cause) |
| AR: ATP6V1B1, ATP6V0A4 (+ sensorineural deafness) | SLE, rheumatoid arthritis |
| CA2 deficiency (mixed proximal-distal) | Obstructive uropathy |
| Pyelonephritis, nephrocalcinosis | |
| Amphotericin B toxicity | |
| Post-renal transplant (CNI toxicity, rejection) |
Presentation
- Hyperchloremic metabolic acidosis (normal anion gap)
- Urine pH persistently > 5.5 (cannot acidify, hallmark finding)
- Hypokalemia - can be profound, causing weakness, paralysis, and even respiratory arrest (notably in Sjogren-associated dRTA) - Brenner & Rector's, p. 2280
- Nephrocalcinosis and nephrolithiasis - occurs in up to 70% of affected individuals; calcium phosphate stones predominate due to hypercalciuria, hypocitraturia, and alkaline urine
- Hypercalciuria - from metabolic acidosis causing bone demineralization and secondary hyperparathyroidism
- Profound hypocitraturia - perhaps the most important stone-risk factor
- Bone disease (rickets in children, osteomalacia in adults)
- Growth retardation, failure to thrive in children
- Urinary anion gap (UAG) is positive (NH4+ excretion is impaired)
Lab Findings Summary
| Parameter | Value |
|---|---|
| Serum pH | Low |
| Serum HCO3- | Low |
| Serum K+ | Low |
| Serum Cl- | High |
| Anion gap | Normal |
| Urine pH | > 5.5 (cannot acidify) |
| UAG | Positive |
| Urine calcium | High |
| Urine citrate | Low |
Ward Management
- Alkali replacement: Oral sodium bicarbonate or potassium citrate 1-2 mEq/kg/day in adults; 3-6 mEq/kg/day in children. Goal serum HCO3- of 22-24 mEq/L. - Goldman-Cecil Medicine
- Potassium citrate is preferred (corrects acidosis AND hypokalemia; citrate is metabolized to bicarbonate and provides K+ simultaneously)
- Acute severe hypokalemia: IV potassium replacement (monitor closely; avoid rapid correction)
- Acute acidemia (pH < 7.2): IV sodium bicarbonate; calculate deficit as: HCO3- deficit = (25 - [HCO3-]) x weight(kg) / 2. Give no more than 50% at a time before recalculation
- Nephrolithiasis: Potassium citrate alkalization reduces stone formation
- Monitor: Serum K+, HCO3-, urine pH, renal function
- Treat underlying cause: e.g., immunosuppression for Sjogren's
Type 2 - Proximal RTA (pRTA)
Pathophysiology
Defective bicarbonate reabsorption in the proximal tubule. The threshold for reabsorption is lowered (from ~22 to ~15 mmol/L). As long as plasma HCO3- is above this lower threshold, bicarbonaturia occurs and urine is alkaline. Once plasma HCO3- falls below the threshold, no further bicarbonate is lost, and urine acidifies normally (urine pH < 5.5 at steady state). This distinguishes it from Type 1. - Campbell Walsh Wein, p. 2689
When HCO3- is given, it is rapidly excreted (fractional excretion of HCO3- rises to 15-20% in pRTA, vs. <3% in dRTA). - Brenner & Rector's
Causes
| Isolated pRTA | Part of Fanconi Syndrome |
|---|---|
| Carbonic anhydrase inhibitors (acetazolamide, topiramate) | Cystinosis (most common hereditary) |
| Sporadic infant form | Wilson's disease |
| AR form: ocular abnormalities, mental retardation | Multiple myeloma (light chains) |
| Heavy metal poisoning (lead, mercury, cadmium) | |
| Tenofovir, ifosfamide, valproic acid | |
| Sjogren syndrome, renal transplant |
Fanconi Syndrome Features: proximal RTA + glycosuria (normal glucose) + hyperaminoaciduria + phosphaturia + hypouricemia + hypercalciuria + hypokalemia. Severe hypophosphatemia with metabolic acidosis should raise this suspicion, especially with AKI. - Brenner & Rector's, p. 2272
Presentation
- Hyperchloremic metabolic acidosis (normal anion gap)
- Urine pH < 5.5 at steady state (can acidify distally when not bicarbonate-wasting)
- Hypokalemia (worsens dramatically with bicarbonate therapy - increased distal HCO3- delivery causes marked kaliuresis)
- No nephrocalcinosis / nephrolithiasis (citrate excretion is relatively normal)
- Metabolic bone disease (rickets, osteomalacia) - from phosphate wasting
- Growth retardation in children
- Features of Fanconi syndrome if present: glycosuria, aminoaciduria, phosphaturia
- UAG: 0 or positive
Ward Management
- Very large doses of bicarbonate required (10-15 mEq/kg/day in children) - because any alkali given is promptly excreted in alkaline urine, and treatment needs to overcome the wasting threshold
- Adults: Treatment often deferred if steady-state acidosis is mild - the stable acidosis paradoxically allows normal net acid excretion; treat symptoms and complications
- Critical caution: Bicarbonate replacement worsens hypokalemia - always co-prescribe potassium chloride supplementation. Monitor K+ closely
- Mixed replacement: Oral citrate + bicarbonate + aggressive KCl supplementation
- Phosphate and Vitamin D: For bone disease and rickets (in Fanconi syndrome): sodium phosphate + calcitriol
- Remove offending agent: Stop tenofovir, ifosfamide, acetazolamide, etc. if drug-induced
- Fractional excretion of HCO3-: Confirm diagnosis - give NaHCO3 load and measure FE-HCO3- (>15% confirms proximal origin)
Type 4 - Hyperkalemic Distal RTA
Pathophysiology
Aldosterone deficiency or aldosterone resistance leads to failure of distal K+ and H+ secretion. Aldosterone normally stimulates H+ secretion by ENaC-mediated Na+ reabsorption (creating negative lumen potential that drives H+ secretion) and directly stimulates ammoniagenesis. Its absence or resistance reduces NH4+ generation and excretion, causing both hyperkalemia and metabolic acidosis. - Campbell Walsh Wein, p. 2689; Tietz Lab Medicine, p. 575
Importantly, these patients can still acidify urine in response to acid challenge (urine pH < 5.5), distinguishing them from Type 1.
Causes
- Hyporeninemic hypoaldosteronism (most common) - diabetic nephropathy, interstitial nephritis, obstructive uropathy, renal transplant, SLE
- Aldosterone deficiency: Addison's disease, adrenalectomy, heparin therapy
- Aldosterone resistance: Pseudohypoaldosteronism (PHA), Gordon syndrome (WNK mutations)
- Drugs: ACE inhibitors, ARBs, NSAIDs (reduce renin), K+-sparing diuretics (amiloride, spironolactone), trimethoprim, calcineurin inhibitors
- CKD with moderate GFR loss (<50 mL/min) - fails to increase aldosterone appropriately. - NKF Primer on Kidney Diseases
Presentation
- Hyperchloremic metabolic acidosis (normal anion gap)
- Hyperkalemia (hallmark - distinguishes from Type 1 and Type 2)
- Urine pH < 5.5 (distal acidification is intact despite impaired NH4+ excretion)
- Mild to moderate metabolic acidosis
- Usually presents with chronic asymptomatic hyperkalemia + mild renal insufficiency; >50% also have hyperchloremic metabolic acidosis
- Strongly associated with diabetic nephropathy - Textbook of Family Medicine
- Stone formation is uncommon
- UAG: positive (NH4+ excretion reduced)
Ward Management
- Treat hyperkalemia first: Dietary potassium restriction + loop diuretics (furosemide) - these also improve urinary acidification without requiring bicarbonate
- Fludrocortisone (mineralocorticoid replacement): 0.1-0.2 mg/day if aldosterone deficiency confirmed; promotes K+ excretion and H+ secretion. Monitor for hypertension and edema
- Sodium bicarbonate or sodium citrate: 0.5-0.8 mEq/kg/day (30-60 mEq/day) - used together with diuretics to aid potassium excretion and correct acidosis. - Goldman-Cecil, p. 1417
- Potassium-binding resins: Sodium polystyrene sulfonate, patiromer, or sodium zirconium cyclosilicate for persistent hyperkalemia
- Avoid/stop offending drugs: ACE inhibitors, ARBs, NSAIDs, K+-sparing diuretics if feasible
- Treat underlying disease: Optimize diabetic nephropathy management (glycemic control, RAAS therapy titration)
- NaHCO3 NOT the primary treatment - correction of hyperkalemia itself usually improves acidification
Comparison Table: All Three Types at a Glance
| Feature | Type 1 (Distal) | Type 2 (Proximal) | Type 4 (Hyperkalemic) |
|---|---|---|---|
| Primary defect | Distal H+ secretion | Proximal HCO3- reabsorption | Aldosterone deficiency/resistance |
| Serum K+ | Low | Low | High |
| Urine pH | > 5.5 (always) | < 5.5 (steady state) | < 5.5 |
| FE-HCO3- | < 3% | > 15% (with HCO3- load) | < 3% |
| Urine AG | Positive | 0 or positive | Positive |
| Nephrocalcinosis/stones | Common (up to 70%) | Rare | Rare |
| Bone disease | Moderate | Severe (Fanconi) | Mild |
| Serum HCO3- | < 15 mEq/L | 15-18 mEq/L (threshold) | 15-20 mEq/L |
| Associated features | Hypercalciuria, hypocitraturia, deafness (AR) | Fanconi syndrome, glycosuria, phosphaturia | Diabetes, CKD, hypertension |
| Alkali dose | 1-2 mEq/kg/day (adults) | 10-15 mEq/kg/day (children) | 0.5-0.8 mEq/kg/day |
| Special treatment | K+ citrate | Vit D + phosphate | Fludrocortisone + loop diuretics |
Ward Diagnostic Approach
Step 1: Confirm normal anion gap metabolic acidosis (AG = Na - [Cl + HCO3-]; normal = 8-12)
Step 2: Check urine pH
- Urine pH > 5.5 in acidosis → Type 1 dRTA
- Urine pH < 5.5 → could be Type 2 (at steady state) or Type 4
Step 3: Check serum potassium
- Hypokalemia → Type 1 or Type 2
- Hyperkalemia → Type 4
Step 4: Urine anion gap (Na + K - Cl): positive = impaired NH4+ excretion (RTA), negative = appropriate NH4+ excretion (diarrhea, GI HCO3- loss)
Step 5: Fractional excretion of HCO3- (with NaHCO3 load):
-
15% → Type 2 (proximal)
- < 3% → Type 1 or 4
Step 6: Check renin/aldosterone if Type 4 suspected; plasma renin activity (low in hyporeninemic hypoaldosteronism)
Sources
- Campbell Walsh Wein Urology, 3-Volume Set, pp. 2688-2689
- Brenner and Rector's The Kidney, 2-Volume Set, pp. 2272-2280
- Goldman-Cecil Medicine, pp. 1413-1418
- Tietz Textbook of Laboratory Medicine, 7th Edition, pp. 562-578
- Tintinalli's Emergency Medicine, p. 2137-2143
This is a shared conversation. Sign in to Orris to start your own chat.