Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching Images
ARDS acute respiratory distress syndrome pathogenesis mechanism diagram
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Searching the Web
ARDS pathogenesis mechanism 2024 2025 update neutrophil alveolar
I now have all the content needed for a comprehensive, well-illustrated answer. Let me compile it.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is the clinical manifestation of severe acute lung injury (ALI) — a syndrome of noncardiogenic pulmonary edema, refractory hypoxemia, and bilateral infiltrates whose pathological substrate is diffuse alveolar damage (DAD).
Overview
The Berlin Definition (2012) requires: acute onset within 1 week of a known insult, bilateral opacities on imaging, PaO₂/FiO₂ ≤ 300 mmHg with PEEP ≥ 5 cmH₂O, and respiratory failure not fully explained by cardiac failure or fluid overload.
Common triggers (>50% of cases from four conditions):
- Sepsis
- Diffuse pulmonary infections (viral, bacterial, Pneumocystis)
- Gastric aspiration
- Severe trauma / head injury
Other causes include pancreatitis, massive transfusion (TRALI), smoke/toxic inhalation, drug overdose, burns, fat embolism, and cardiopulmonary bypass.
Pathogenesis: Step-by-Step
1. Initiating Insult → Alveolar-Capillary Injury
ARDS is initiated by injury to pneumocytes (alveolar epithelium) and/or pulmonary endothelium, either:
- Direct (pulmonary route): aspiration, pneumonia, inhalation injury → direct pneumocyte damage sensed by resident alveolar macrophages
- Indirect (systemic route): sepsis, trauma, pancreatitis → circulating mediators (endotoxin, cytokines) activate pulmonary endothelium from the vascular side
2. Endothelial Activation and Leukocyte Recruitment
Resident alveolar macrophages are the sentinel immune cells. Activated by DAMPs (from pneumocyte injury) or PAMPs (from infection/sepsis), they secrete:
- TNF-α, IL-1β, IL-8, IL-6 — pro-inflammatory cytokines
- Platelet-activating factor (PAF) and chemokines
These cytokines act on the neighboring capillary endothelium, inducing expression of adhesion molecules (ICAM-1, E-selectin, P-selectin), procoagulant proteins, and further chemokines — priming it for neutrophil recruitment.
3. Neutrophil Sequestration and Degranulation (Central Amplifying Step)
Circulating neutrophils adhere to the activated endothelium, then migrate (diapedese) into the interstitium and alveolar spaces. Once there, they:
- Degranulate → release proteases (elastase, matrix metalloproteinases), myeloperoxidase
- Release reactive oxygen species (ROS) → oxidative membrane damage
- Secrete more cytokines (IL-1, TNF) → amplifying feedback loop
- Form neutrophil extracellular traps (NETs) → direct physical damage to alveolar structures and promotion of microvascular thrombosis
This neutrophil-driven injury is self-perpetuating: endothelial damage → more cytokines → more neutrophil recruitment.
4. Disruption of the Alveolar-Capillary Barrier
The combined endothelial and epithelial injury makes capillaries massively permeable:
- Protein-rich fluid floods from capillaries into the interstitium and alveolar spaces → noncardiogenic pulmonary edema
- Type I pneumocytes (which cover ~95% of the alveolar surface) are particularly vulnerable and undergo necrosis → loss of the epithelial barrier
- Type II pneumocytes, which produce surfactant, are damaged → surfactant is both reduced in quantity and inactivated by edema proteins → alveolar collapse (atelectasis) and ↑ surface tension
5. Hyaline Membrane Formation (Hallmark of DAD)
Over 24–72 hours, the protein-rich intra-alveolar exudate — composed of plasma proteins, fibrin, and necrotic cell debris — condenses against the denuded alveolar walls to form hyaline membranes. These eosinophilic, linear aggregates are the diagnostic histological feature of DAD/ARDS.
Simultaneously, fibrin deposits within capillary lumens contribute to microvascular thrombosis, worsening perfusion.
6. Physiologic Consequences → Hypoxemia
The mechanical result of these changes is:
- V/Q mismatch: poorly ventilated, consolidated regions remain perfused → intrapulmonary shunt → refractory hypoxemia unresponsive to supplemental O₂
- Reduced lung compliance: stiff, edematous lung (the "baby lung" concept — only small, non-dependent zones remain recruitable)
- Pulmonary hypertension: from hypoxic vasoconstriction, microvascular thrombosis, and mediator-driven vasoconstriction → increased RV afterload
- The lesion distribution is non-uniform: dependent zones consolidate and flood (atelectatic/recruitable), while non-dependent zones may retain near-normal compliance
Histological Phases
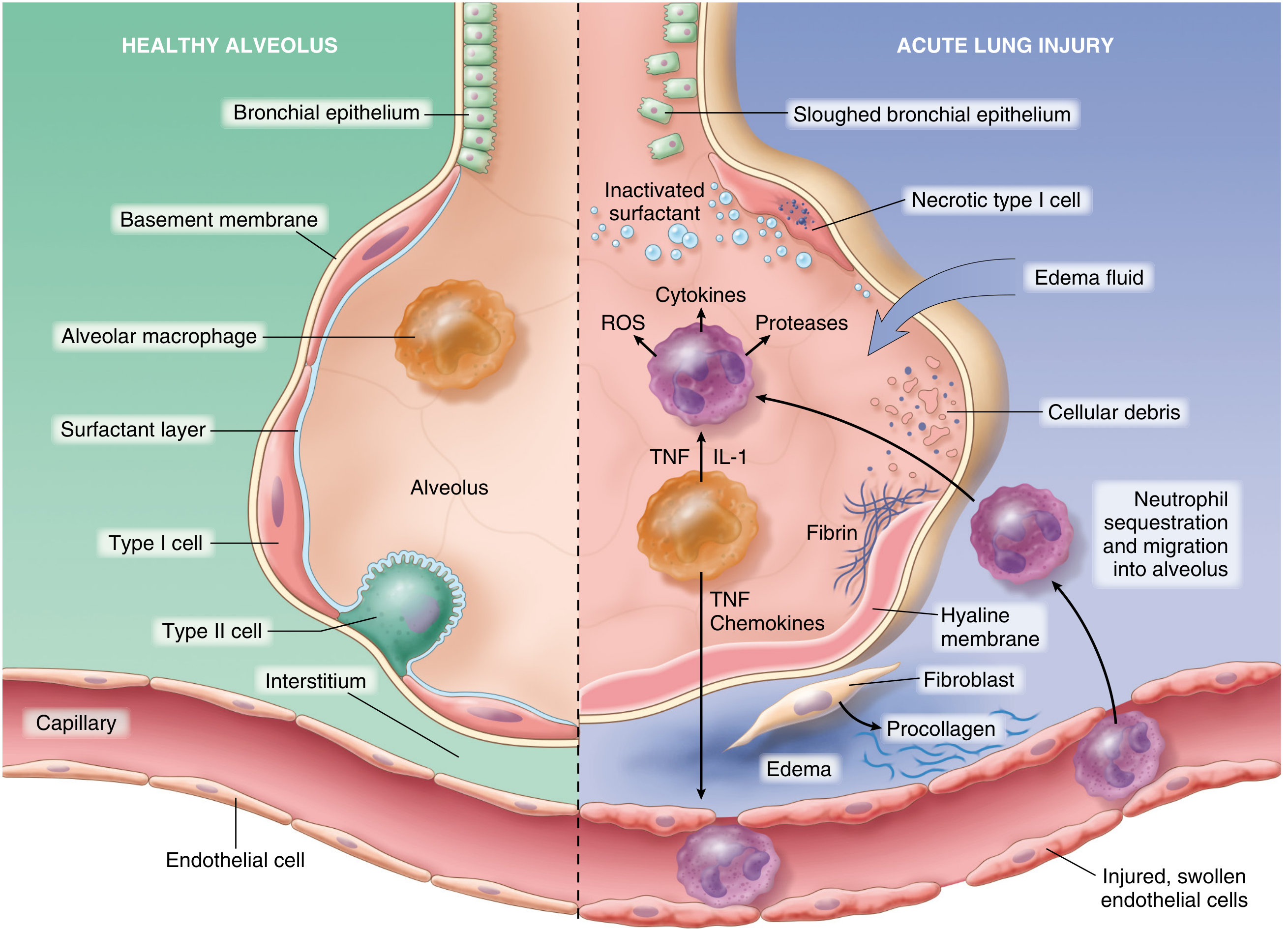
Fig. 15.3 — Normal alveolus (left) vs. injured alveolus in acute lung injury/ARDS. Key components: TNF/IL-1 from macrophages activate endothelium → neutrophil sequestration → ROS, proteases, cytokines → edema, hyaline membrane formation, surfactant inactivation. (Robbins Pathology / Matthay et al.)
| Phase | Timing | Histology |
|---|---|---|
| Exudative (acute) DAD | Days 1–7 | Interstitial edema, type I pneumocyte necrosis, neutrophilic infiltrate, hyaline membranes, alveolar flooding |
| Proliferative (organizing) DAD | Days 7–21 | Type II pneumocyte hyperplasia (re-epithelialization), fibroblast influx, granulation tissue plugs, residual hyaline membranes |
| Fibrotic | Weeks–months | Variable: complete resolution OR persistent fibrosis, alveolar simplification, impaired function |
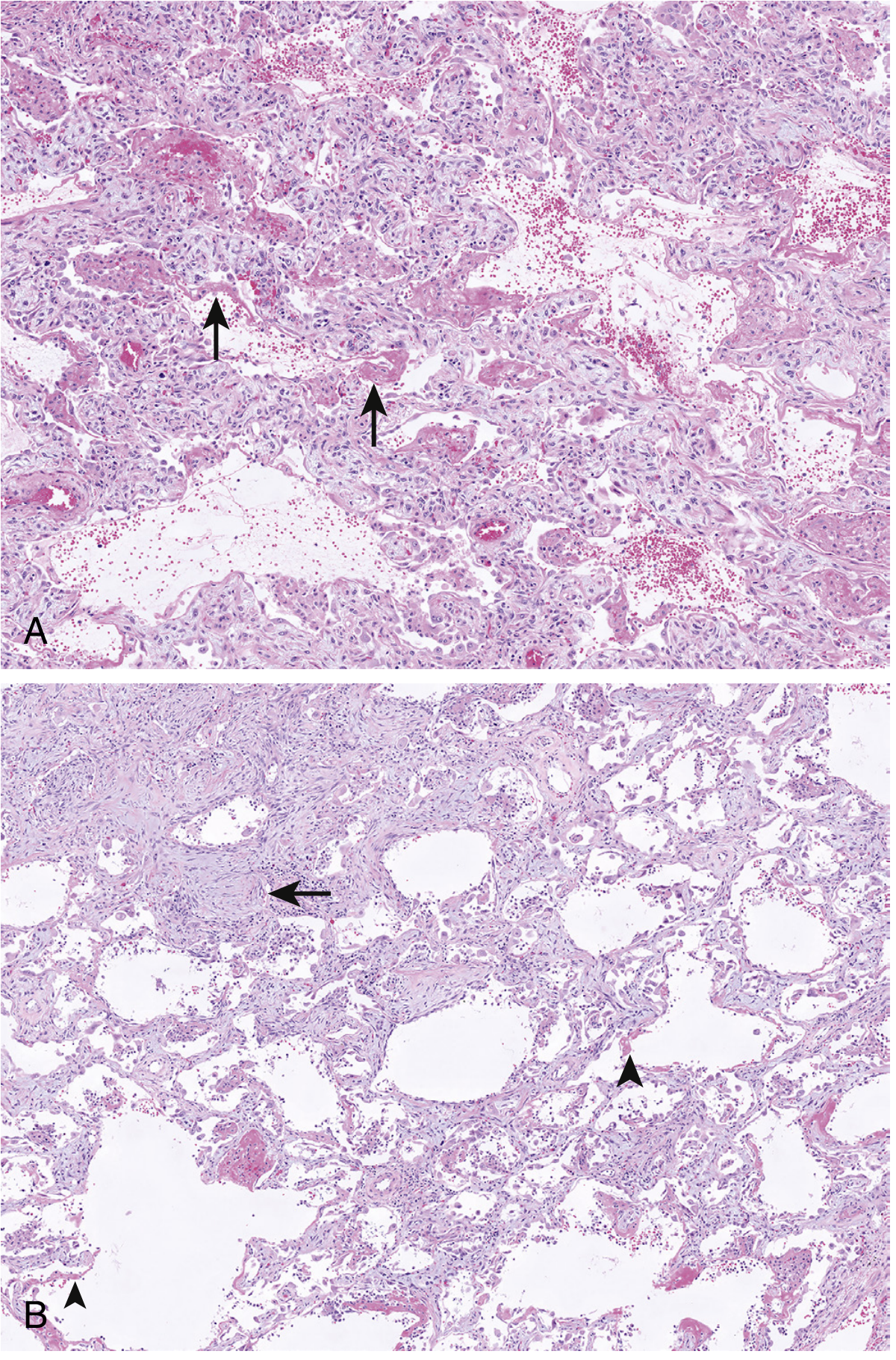
Figure 22.6 — DAD histology. (A) Acute phase: interstitial edema, type II cell hyperplasia, hyaline membranes (arrows). (B) Organizing phase: granulation tissue plugs (arrow), residual hyaline membranes (arrowheads). H&E ×100. — Murray & Nadel's Textbook of Respiratory Medicine
Resolution and Repair
If the triggering insult abates, repair depends on:
- Macrophage clearance of intra-alveolar debris
- Release of TGF-β and PDGF → fibroblast proliferation and collagen deposition (risk of fibrosis)
- Type II pneumocyte proliferation → differentiate into type I cells, restoring the epithelial lining
- Endothelial regeneration from uninjured capillary cells
In patients who survive, the fibroproliferative phase (weeks 2–4) carries its own risk — high fibroblast activity can produce progressive pulmonary fibrosis. Complete recovery is possible but may take months; some patients sustain lasting impairment.
Summary of Key Mediators
| Mediator | Source | Role in ARDS |
|---|---|---|
| TNF-α, IL-1β | Macrophages | Endothelial activation, neutrophil priming |
| IL-8 (CXCL8) | Macrophages, epithelium | Neutrophil chemotaxis |
| ROS | Neutrophils | Membrane lipid peroxidation, cell death |
| Elastase, MMPs | Neutrophils | ECM degradation, barrier disruption |
| NETs | Neutrophils | Direct lung injury, microvascular thrombosis |
| PAF | Macrophages, endothelium | Neutrophil activation, vasoconstriction |
| Phospholipase A2 | Pancreatic (in pancreatitis-ARDS) | Surfactant degradation |
| TGF-β, PDGF | Macrophages (repair phase) | Fibroblast activation → fibrosis |
Sources: Robbins & Cotran Pathologic Basis of Disease, 10th ed. (p. 632); Murray & Nadel's Textbook of Respiratory Medicine, 7th ed. (pp. 566, 2159); Goldman-Cecil Medicine, 27th ed. (p. 1066).
This is a shared conversation. Sign in to Orris to start your own chat.