Dmd
Reading File
Finding Sources
Searching PubMed
"Duchenne muscular dystrophy"[MeSH Terms] AND treatment
Reading File
Reading File
Duchenne Muscular Dystrophy (DMD)
Overview
DMD is the most common inherited muscular dystrophy, affecting approximately 1 in 3,500-5,000 male births. It is a fatal X-linked recessive disorder caused by frameshift mutations in the DMD gene at locus Xp21.2, which encodes dystrophin - a large structural protein that mechanically links the sarcomere (via actin) to the sarcolemma and extracellular matrix. Without functional dystrophin, repeated muscle contraction causes a calcium leak across the membrane, triggering necrosis and progressive replacement of muscle by fibroadipose tissue.
About 1/3 of cases arise from de novo mutations with no family history.
Genetics & Molecular Pathology
- Gene: DMD (Xp21.2) - one of the largest human genes (~2.4 Mb)
- Mutation types: Frameshift deletions/duplications (~70%), point mutations (~30%)
- Result: Complete absence of dystrophin protein (vs. partial loss in the milder Becker MD)
- The reading frame rule: Out-of-frame mutations = DMD (severe); in-frame mutations = Becker MD (milder)
- Female carriers: Usually asymptomatic, but up to 20% can show muscle weakness or dilated cardiomyopathy (DCM) due to skewed X-inactivation (lyonization)
Clinical Features
| Stage | Age | Features |
|---|---|---|
| Early | 2-5 years | Delayed motor milestones, frequent falls, Gowers' sign, pseudohypertrophy of calves |
| Progressive | 5-12 years | Proximal muscle weakness (lower > upper limbs), waddling gait, Trendelenburg sign |
| Wheelchair-bound | 10-15 years | Loss of ambulation; scoliosis, joint contractures develop |
| Late | By ~20 years | Kyphoscoliosis, severe respiratory compromise, cardiomyopathy |
Associated features: Lower IQ (~30%), learning disorders, autism spectrum disorder, ADHD (the degree of cognitive impairment may correlate with mutation location in the gene).
Cardiac Involvement
- Dilated cardiomyopathy (DCM) is the hallmark - caused by progressive cardiac fibrosis and left ventricular dilation
- Rhythm/conduction abnormalities are common
- Cardiorespiratory failure is the primary cause of death
- Some patients develop isolated X-linked DCM (OMIM #302045) without severe skeletal muscle disease
Diagnosis
- Serum CK: Elevated 20-100x above normal (markedly elevated even presymptomatically)
- Genetic testing (DNA analysis): Positive in ~90-95% of patients - first-line confirmation
- Muscle biopsy (if genetic testing negative, ~5% of cases):
- Histology: Variation in fiber size, necrosis, inflammation, fibrosis, regeneration
- Immunohistochemistry (IHC): Complete/near-complete absence of dystrophin (carboxy-terminal antigens)
- "Revertant fibers" (small clusters retaining dystrophin) may be seen
- Newborn screening: Elevated CK can be detected from blood spots; two-tiered CK + DNA programs have been piloted (e.g., Ohio) but are not yet standard in the US
Treatment
Management requires a multidisciplinary team (neurology, cardiology, pulmonology, orthopedics, physiotherapy, psychosocial support, nutrition).
Corticosteroids (mainstay)
- Prednisone 0.75 mg/kg/day - prolongs ambulation, helps respiratory function, slows scoliosis progression
- Deflazacort 0.9 mg/kg/day - similar efficacy to prednisone with less weight gain; FDA-approved for DMD
Exon-Skipping Therapies (FDA-conditionally approved)
These phosphorodiamidate morpholino oligomers (PMOs) restore the reading frame by skipping specific exons:
| Drug | Target exon | Dose |
|---|---|---|
| Eteplirsen | Exon 51 | 30 mg/kg IV weekly |
| Golodirsen | Exon 53 | 30 mg/kg IV weekly |
| Viltolarsen | Exon 53 | 80 mg/kg IV weekly |
| Casimersen | Exon 45 | 30 mg/kg IV weekly |
These apply only to patients with eligible mutations; expert neurologic consultation required for selection.
Cardiac Management
- ACE inhibitors - slow progression of myocardial fibrosis; started early even before symptomatic cardiomyopathy
- Beta-blockers and diuretics for established DCM
Respiratory Management
- Regular pulmonary function monitoring
- Noninvasive ventilation (BiPAP/NIV) as respiratory function declines
Orthopedic Management
- Physiotherapy to prevent contractures
- Monitoring for scoliosis; spinal fusion when indicated
- Timely provision of wheelchairs and assistive devices
Prognosis
Without treatment, most patients die in their late teens to 20s from cardiorespiratory failure. With modern multidisciplinary care, corticosteroids, and ventilatory support, survival into the 30s and beyond is increasingly achievable.
Sources: Goldman-Cecil Medicine (International Edition); Tietz Textbook of Laboratory Medicine, 7th Ed.
Management of status asthmaticus
Reading File
Finding Sources
Searching PubMed
"status asthmaticus"[MeSH Terms] AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Management of Status Asthmaticus
Status asthmaticus is acute severe asthma that does not respond to usual doses of inhaled bronchodilators and corticosteroids. It is a life-threatening emergency requiring rapid, aggressive intervention.
NAEPP ED/Hospital Management Algorithm
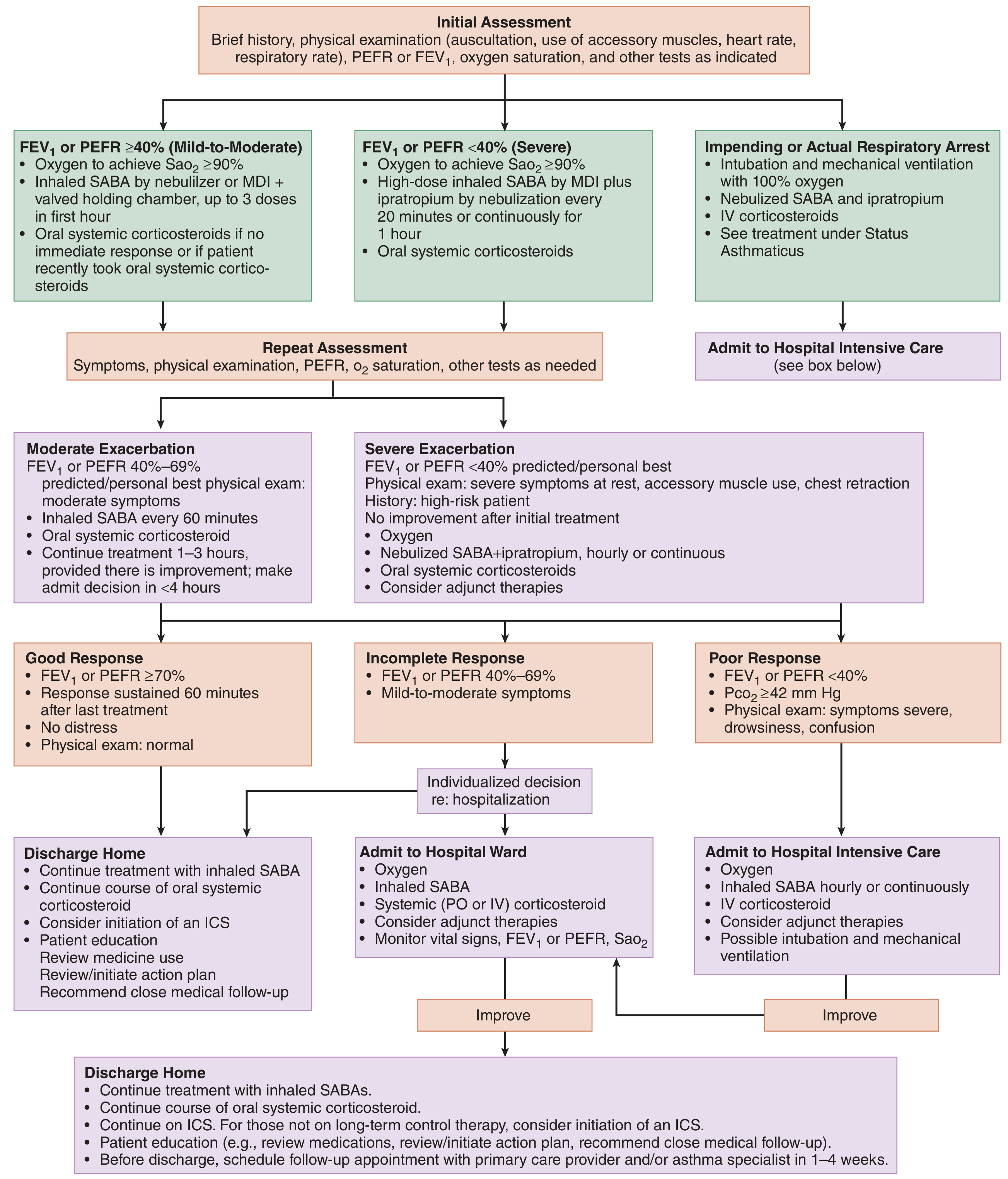
Step 1 - Initial Assessment
Clinical features: Hypoxemia, tachypnea, tachycardia, accessory muscle use, wheezing (may be absent when airflow is severely reduced - "silent chest" = ominous sign), confusion/drowsiness.
Severity classification (by FEV1 or PEFR):
| Category | FEV1/PEFR | Features |
|---|---|---|
| Mild-Moderate | ≥40% | Minimal accessory muscle use |
| Severe | <40% | Accessory muscle use, chest retraction, rest symptoms |
| Impending arrest | Near-zero airflow | Silent chest, cyanosis, altered sensorium |
Key investigations: ABG (PaCO2 rising toward normal/above normal = impending fatigue), SpO2, peak flow or spirometry, CXR (to exclude pneumothorax, pneumonia).
Note: In early severe asthma, PaCO2 is low (hyperventilation). A normal or rising PaCO2 signals respiratory muscle fatigue - prepare for intubation.
Step 2 - Immediate Treatment
1. Oxygen
- Target SpO2 ≥90% (≥95% in pregnancy, children)
- Controlled O2 via mask/nasal prongs; avoid high-flow that may delay recognition of deterioration
2. Short-Acting Beta-2 Agonists (SABA) - First-line, most important
| Setting | Albuterol dose | Route |
|---|---|---|
| Mild-Moderate | 2.5-5 mg Q20 min x 3 doses | Nebulizer |
| Mild-Moderate | 4-8 puffs Q20 min | MDI + spacer |
| Severe (adult) | Continuous nebulization | 10-15 mg/hr continuous |
| Severe (pediatric) | 0.5 mg/kg/hr (max 30 mg/hr) | Continuous nebulization |
- MDI + spacer is preferred when the patient can cooperate - equal or greater efficacy, fewer side effects, shorter hospital stay
- Levalbuterol has no proven advantage over racemic albuterol and costs significantly more
3. Ipratropium Bromide (Anticholinergic)
- Add to SABA for severe exacerbations
- Dose: 0.5 mg nebulized Q20 min x 3 doses (or 4-8 puffs MDI)
- Provides additive bronchodilation by blocking muscarinic-mediated bronchoconstriction
- Note: No proven additional benefit in the inpatient setting (use in ED/initial phase)
4. Systemic Corticosteroids - Start early
| Drug | Adult dose | Pediatric dose |
|---|---|---|
| Prednisone/Prednisolone | 40-60 mg PO daily x 5-7 days | 2 mg/kg/day PO (max 60 mg) |
| Methylprednisolone | 60-80 mg IV Q6-12 hr | Q6-12 hr (max 60-80 mg/day by age) |
| Dexamethasone (pediatric) | - | 0.6 mg/kg/day x 1-2 days (max 16 mg) |
- No proven advantage for IV over oral when GI absorption is intact
- No proven benefit from higher doses in severe exacerbations
- Courses <7 days: no taper needed
- Inhaled corticosteroids can be started at any point during treatment
Step 3 - Adjunct Therapies (for Refractory Cases)
Magnesium Sulfate
- IV MgSO4: 1-2 g IV over 20-30 min (pediatric: 25-75 mg/kg max 2 g)
- Indicated for FEV1/PEFR <25% predicted or inadequate response to bronchodilators
- Mechanism: inhibits smooth muscle contraction (calcium antagonism)
- Nebulized MgSO4: 95-384 mg in sterile water; effective as adjunct after aggressive beta-agonist/steroid therapy
- Monitor: BP and deep tendon reflexes during infusion (hypotension and neuromuscular blockade are rare at these doses)
Epinephrine / Terbutaline (Parenteral)
- For severe exacerbations with minimal air entry where inhaled therapy is ineffective
- Epinephrine: 0.01 mg/kg IM (1:1000, max 0.5 mg) Q15-20 min x 3 doses
- Terbutaline SC: 0.01 mg/kg SC (max 0.25 mg) Q20 min x 3 doses; or IV continuous 0.2-5 mcg/kg/min
- Terbutaline IV may reduce the need for mechanical ventilation
Ketamine
- 1-2 mg/kg IV load + 1 mg/kg/hr infusion, titrated to effect
- Bronchodilatory properties via catecholamine release and direct smooth muscle relaxation
- Useful for preintuabtion sedation and for intubated patients not improving on MV
- Preferred induction agent for RSI in status asthmaticus
Heliox (Helium-Oxygen mixture, typically 70:30)
- Reduces turbulent airflow through narrowed airways; reduces work of breathing
- Useful as a bridge while other therapies take effect
- Most beneficial when helium concentration is maximized (limits FiO2 to 30% - avoid if hypoxic)
Aminophylline (controversial, less used now)
- 6 mg/kg IV load over 20 min, then 0.5-1.2 mg/kg/hr infusion
- Narrow therapeutic window, multiple drug interactions, cardiac toxicity risk
- Generally not first-line; used only when other therapies fail
Step 4 - Ventilatory Support
Non-Invasive Ventilation (BiPAP/NIV)
- Use for preoxygenation before intubation
- Reduces work of breathing, may temporarily stave off respiratory arrest
- Reassurance and coaching are key - anxious patients may resist mask
Mechanical Ventilation - Indications
- Impending or actual respiratory arrest
- Altered consciousness, exhaustion, cyanosis despite O2
- Rising PaCO2 with worsening acidosis
RSI Protocol for Status Asthmaticus
| Time | Action |
|---|---|
| -10 min | Preparation, IV access, monitoring |
| -5 min | Preoxygenate: continuous albuterol nebulizer + 100% O2 via NRB or BiPAP |
| -3 min | IV epinephrine or SC terbutaline; albuterol 2.5 mg nebulized |
| 0 | Ketamine 1.5 mg/kg IV + Succinylcholine 1.5 mg/kg IV |
| +30 s | Positioning |
| +45 s | Laryngoscopy + intubation; confirm with end-tidal CO2 |
| +2 min | Sedation/analgesia; NMBA only if needed after adequate sedation |
Post-Intubation Ventilator Strategy ("Controlled Hypoventilation")
- Low RR (8-12/min) with low tidal volume (6-8 mL/kg) - allow adequate exhalation time
- High inspiratory flow rate to maximize expiratory time (I:E ratio 1:3 to 1:5)
- Permissive hypercapnia acceptable - avoid auto-PEEP (breath stacking)
- Auto-PEEP causes barotrauma and hemodynamic compromise
- Long-term NMBA may be needed to reduce oxygen consumption and ventilator dysynchrony
Extracorporeal Life Support (ECMO)
- Last-resort option for refractory cases with life-threatening hypoxia or hemodynamic compromise
Step 5 - Agents of No Proven Benefit (avoid)
- Sodium bicarbonate (does not improve outcomes)
- Antibiotics (unless clear evidence of bacterial infection - most exacerbations are viral)
- IV beta-agonists (no advantage over inhaled; increased systemic side effects)
- Lidocaine for RSI (no high-quality evidence)
Discharge Criteria & Plan
- FEV1/PEFR ≥70% and sustained 60 min after last treatment
- SpO2 stable, no distress, normal exam
- Discharge medications: SABA PRN, oral corticosteroids (prednisone 40 mg x 5-7 days), initiate/continue ICS
- Follow-up: within 1-4 weeks with PCP or asthma specialist
- Provide written asthma action plan and peak flow diary
Recent evidence note: A 2024 systematic review (PMID 39402635, Crit Care) found that inhaled sevoflurane is an emerging option for adults with refractory status asthmaticus in the ICU, with bronchodilatory properties and favorable safety profile - though it remains outside standard practice guidelines.
Sources: Tintinalli's Emergency Medicine, 9th Ed.; Rosen's Emergency Medicine, 9th Ed.; Harriet Lane Handbook, 23rd Ed. (pediatric dosing)
Floppy child
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive data. Let me compile the full answer.
The Floppy Child (Infantile Hypotonia)
Definition
A "floppy child" describes an infant or young child with decreased muscle tone (hypotonia) - manifesting as reduced resistance to passive movement and abnormal posturing. It is a common but diagnostically challenging presentation in pediatric neurology.
Clinical Recognition
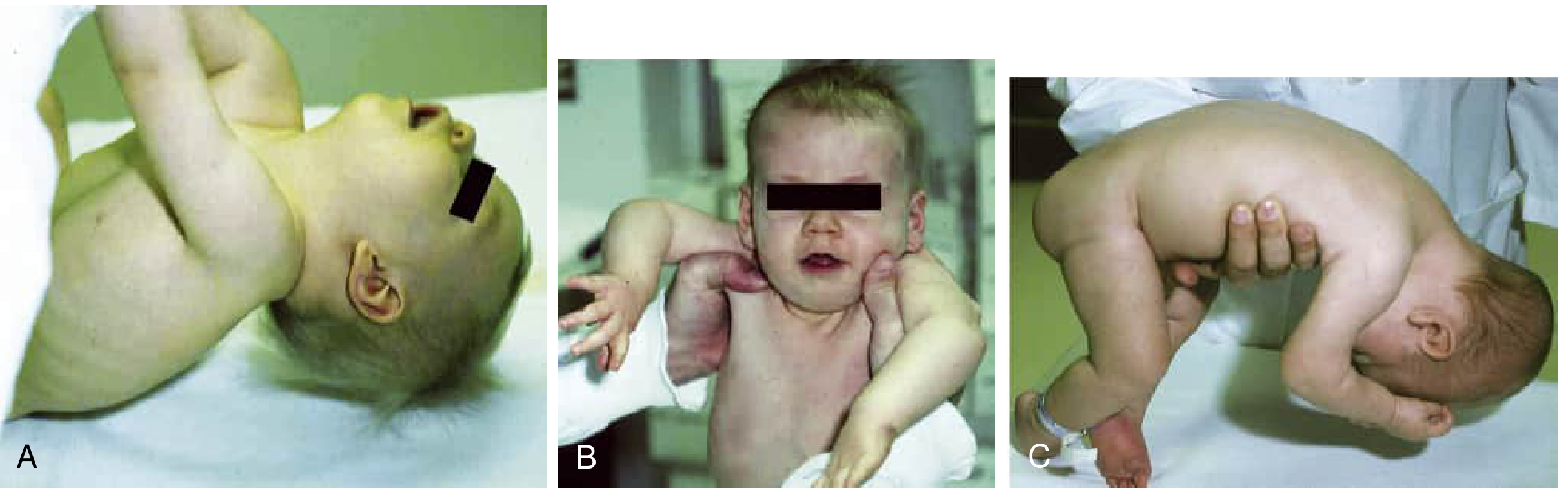
Fig. A - Abnormal traction response with excessive head lag. B - Vertical suspension showing "slip-through" (shoulders elevate, arms extend, infant slips through examiner's hands). C - Horizontal suspension with head and limbs hanging limply.
Key physical signs:
- "Frog-leg" posture in supine - legs abducted, externally rotated, hips and knees flexed with laxity
- Excessive head lag on traction (pull-to-sit) - head falls back completely
- Slip-through sign in vertical suspension - infant slides through examiner's hands
- Draping in horizontal (ventral) suspension - head and limbs hang limply
- Increased passive range of motion at ankles and hips
- Weak cry, poor suck, feeding difficulties
Step 1 - The Critical Branch Point: Central vs. Peripheral
60-80% of infantile hypotonia is CENTRAL (brain/spinal cord); 15-30% is PERIPHERAL (motor unit)
| Feature | Central (UMN) Hypotonia | Peripheral (LMN) Hypotonia |
|---|---|---|
| Alertness/consciousness | Encephalopathy, seizures, altered consciousness | Usually alert and awake |
| Tendon reflexes | Normal or increased (may be decreased early) | Absent or markedly reduced |
| Weakness | Mild relative to degree of hypotonia | Prominent weakness, proportional to hypotonia |
| Primitive reflexes | Often preserved or exaggerated | Depressed or absent |
| Dysmorphic features | Common (chromosomal, CNS malformations) | Less common (except specific syndromes) |
| CK | Usually normal | Elevated in myopathies, some SMA |
| Fasciculations | Absent | Present in anterior horn cell disease (SMA) |
| Sensory deficits | Rare | Present in neuropathies |
Differential Diagnosis - Full Classification
A. CENTRAL CAUSES (~60-80%)
1. Hypoxic-Ischemic Encephalopathy (HIE)
- Most common acquired cause
- History of perinatal distress, birth asphyxia
- Encephalopathy + seizures + multiorgan dysfunction
2. Chromosomal / Genetic Syndromes
- Down syndrome (Trisomy 21) - most common chromosomal cause; characteristic facies, hypotonia, Brushfield spots
- Prader-Willi syndrome - severe neonatal hypotonia, poor feeding in infancy → insatiable appetite/obesity later; hypoplastic genitalia, intellectual impairment
- Angelman syndrome - hypotonia + seizures + absent speech + inappropriate laughter
3. Structural Brain Malformations
- Lissencephaly, holoprosencephaly, polymicrogyria
- Cerebellar hypoplasia
4. Inborn Errors of Metabolism
- Present after a symptom-free interval once feeding begins (toxic intermediate accumulation)
- Amino acidopathies, organic acidurias, urea cycle defects
- Congenital lactic acidosis - mitochondrial disease, pyruvate metabolism defects
- Congenital disorders of glycosylation (CDG) - hypotonia + cerebellar hypoplasia + coagulopathy + inverted nipples + abnormal fat distribution
- Pompe disease (Glycogen storage type II) - cardiomegaly + hypotonia + macroglossia
5. Spinal Cord Injury
- Perinatal - difficult breech delivery or hypoxic-ischemic injury
- Clues: absent pain response below level, sphincter dysfunction, priapism
B. PERIPHERAL CAUSES (~15-30%)
Anterior Horn Cell (Spinal Motor Neuron)
Spinal Muscular Atrophy (SMA) - Werdnig-Hoffmann Disease
- Most important peripheral cause - autosomal recessive, SMN1 deletion (Xp21.2)
- SMA Type 0: weakness at birth, arthrogryposis, respiratory failure in early infancy
- SMA Type 1 (Werdnig-Hoffmann): onset <6 months, proximal > distal weakness, fasciculations of tongue, abdominal breathing (diaphragm relatively spared), absent reflexes
- CK may be mildly elevated (<1000 U/L)
- EMG: acute and chronic denervation
- Treatment is urgent: nusinersen (Spinraza - intrathecal antisense oligonucleotide), onasemnogene abeparvovec (Zolgensma - gene replacement), risdiplam (Evrysdi - oral); benefit decreases with delay
Peripheral Neuropathy
- Rare cause; includes congenital hypomyelinating neuropathy and Dejerine-Sottas disease
- Reflexes disproportionately absent relative to weakness (demyelinating pattern)
- NCS: markedly slowed conduction velocities
Neuromuscular Junction (NMJ)
Neonatal Myasthenia Gravis
- Transient - maternal antibodies (anti-AChR) cross placenta
- Mother has myasthenia gravis; resolves within weeks
- Fatigable weakness, ptosis, poor suck
Congenital Myasthenic Syndromes
- Genetic (not antibody-mediated); persistent
- Various mutations in NMJ proteins (AChR subunits, rapsyn, ColQ, etc.)
Infant Botulism - IMPORTANT to consider
- Ingestion of Clostridium botulinum spores (honey, soil)
- Age: 1-6 months typically
- Descending paralysis: constipation → poor feeding → hypotonia → respiratory failure
- Dilated, poorly reactive pupils; absent gag; repetitive nerve stimulation shows incremental pattern
- Treatment: BabyBIG (human botulism immune globulin)
Muscle Disorders
Congenital Myopathies
- Generally normal or mildly elevated CK
- Diagnosis by muscle biopsy + genetic testing
| Type | Histology | Gene |
|---|---|---|
| Central core disease | Central cores (absent mitochondria, no oxidative activity) | RYR1 |
| Nemaline myopathy | Nemaline (rod) bodies | Multiple (NEB, ACTA1, TPM2, TPM3) |
| Centronuclear myopathy | Internal nuclei | MTM1, DNM2, BIN1 |
Congenital Muscular Dystrophies
- Elevated CK, early onset hypotonia and contractures
- Merosin-deficient CMD - absent laminin-α2 on biopsy, white matter changes on MRI
- Ullrich CMD - collagen VI deficiency; joint hypermobility distally + contractures proximally
- Syndromic CMD (cobblestone lissencephaly group): Fukuyama CMD, Walker-Warburg syndrome, Muscle-Eye-Brain disease - severe myopathy + CNS malformation + ocular defects
Congenital Myotonic Dystrophy
- Maternal inheritance (trinucleotide repeat expansion in DMPK gene)
- ~25% of infants of affected mothers; mother's diagnosis may be unrecognized
- Facial diplegia, hypotonia, respiratory distress → later myotonia in adolescence
Diagnostic Workup
Tier 1 - All hypotonic infants
- Detailed history: pregnancy, perinatal period, family history, consanguinity
- Careful physical and neurological examination
- Chromosomal microarray (first-line genetic test)
- Serum CK (markedly elevated → muscular dystrophy/myopathy; normal → central or some myopathies)
- Metabolic screen: blood ammonia, plasma amino acids, acylcarnitine profile, urine organic acids, lactate/pyruvate, glucose
Tier 2 - Based on localization
| Suspected localization | Test |
|---|---|
| CNS | Brain MRI (T1/T2, DWI, MRS) |
| Anterior horn cell | SMN1 deletion analysis (SMA); EMG/NCS |
| NMJ | Repetitive nerve stimulation; anti-AChR Ab (neonatal MG); stool culture (botulism) |
| Neuropathy | NCS (slowed velocities in demyelinating neuropathy) |
| Myopathy | Muscle biopsy (histology + IHC + EM); gene panel |
Tier 3 - Targeted testing
- EEG if seizures suspected
- Echocardiogram if cardiomyopathy suspected (Pompe disease)
- Ophthalmology for structural CNS/eye diseases
- Thyroid function (hypothyroidism - treatable cause)
A systematic diagnostic approach yields a specific diagnosis in up to 85% of cases.
Management Principles
- SMA is a medical emergency - start genetic testing immediately; disease-modifying therapy is time-sensitive
- Metabolic emergencies: hold feeds, start IV glucose, investigate and treat empirically for sepsis while awaiting cultures
- Neonatal MG: resolves spontaneously; neostigmine if feeding/respiratory support needed
- Infant botulism: BabyBIG IV (human botulism immune globulin); ICU/ventilatory support
- Multidisciplinary care: neurology, genetics, physiotherapy, occupational therapy, speech/feeding therapy, respiratory support
- Arthrogryposis: physiotherapy, splinting, orthopaedic referral for contractures
- Genetic counselling for familial conditions (SMA, congenital myopathies, CMDs)
Sources: Bradley and Daroff's Neurology in Clinical Practice; Adams and Victor's Principles of Neurology, 12th Ed.; Harriet Lane Handbook, 23rd Ed.
This is a shared conversation. Sign in to Orris to start your own chat.