Throud disorders , mainly hyper and hypo
Reading File
Finding Sources
Searching PubMed
"thyroid disorders" AND "hyperthyroidism OR hypothyroidism"
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Finding Sources
I now have comprehensive material from Harrison's 22E (2025) and other authoritative sources. Let me compile the full answer.
Thyroid Disorders: Hyperthyroidism and Hypothyroidism
Overview of Thyroid Hormones
The thyroid gland produces thyroxine (T4) and triiodothyronine (T3), regulated by TSH from the pituitary (which is itself driven by TRH from the hypothalamus). T4 is converted peripherally to the more active T3. Both hormones affect metabolism, cardiac function, neuromuscular activity, and virtually every organ system.
HYPERTHYROIDISM (Thyrotoxicosis)
Thyrotoxicosis = state of thyroid hormone excess (not synonymous with hyperthyroidism, which specifically means excessive thyroid gland function).
Causes
Thyrotoxicosis WITH hyperthyroidism (high gland output):
| Cause | Notes |
|---|---|
| Graves' disease | 60-80% of all thyrotoxicosis; autoimmune (TRAb stimulate TSH-R) |
| Toxic multinodular goiter (MNG) | Common in elderly, iodine-deficient areas |
| Solitary toxic adenoma | Autonomous nodule, usually >3 cm |
| TSH-secreting pituitary adenoma | Rare; TSH is NOT suppressed |
| Activating TSH-R or Gsa mutations | McCune-Albright syndrome |
| Gestational thyrotoxicosis | hCG cross-reacts with TSH receptor |
Thyrotoxicosis WITHOUT hyperthyroidism (destructive - hormone release, not excess production):
- Subacute (de Quervain's) thyroiditis
- Silent/postpartum thyroiditis
- Amiodarone, cytokines, immune checkpoint inhibitors
- Thyrotoxicosis factitia (excess exogenous thyroid hormone)
(Harrison's 22E, Table 396-1)
Graves' Disease - Deep Dive
Epidemiology: Affects up to 2% of women; 1/10th as common in men. Peak age 20-50 years.
Pathogenesis: TSH receptor antibodies (TRAb, also called TSI) stimulate the TSH-R, causing unregulated thyroid hormone production. Genetic susceptibility involves HLA-DR, CTLA-4, CD25, CD40, PTPN22 polymorphisms. Concordance in monozygotic twins is 20-30%, suggesting strong environmental contribution. Stress, smoking, and iodine excess are key environmental triggers.
The triad of Graves':
- Thyrotoxicosis (diffuse goiter)
- Ophthalmopathy (proptosis, lid lag, lid retraction, periorbital edema)
- Dermopathy (pretibial myxedema - rare, ~1-2%)
Clinical Features of Hyperthyroidism
| System | Symptoms | Signs |
|---|---|---|
| General | Heat intolerance, weight loss despite good appetite, fatigue | Hyperthermia, sweating |
| Cardiovascular | Palpitations, dyspnea | Tachycardia, atrial fibrillation, wide pulse pressure |
| Neuromuscular | Tremor, anxiety, irritability, insomnia | Fine tremor, proximal muscle weakness, hyperreflexia |
| GI | Increased bowel frequency, diarrhea | - |
| Reproductive | Oligomenorrhea, infertility | Gynecomastia (men) |
| Skin/Hair | - | Warm, moist skin; fine hair; onycholysis (Plummer's nails) |
| Eyes | Gritty sensation, double vision | Proptosis, lid lag, chemosis (in Graves') |
Diagnosis of Hyperthyroidism
The diagnostic algorithm from Harrison's 22E is shown below:
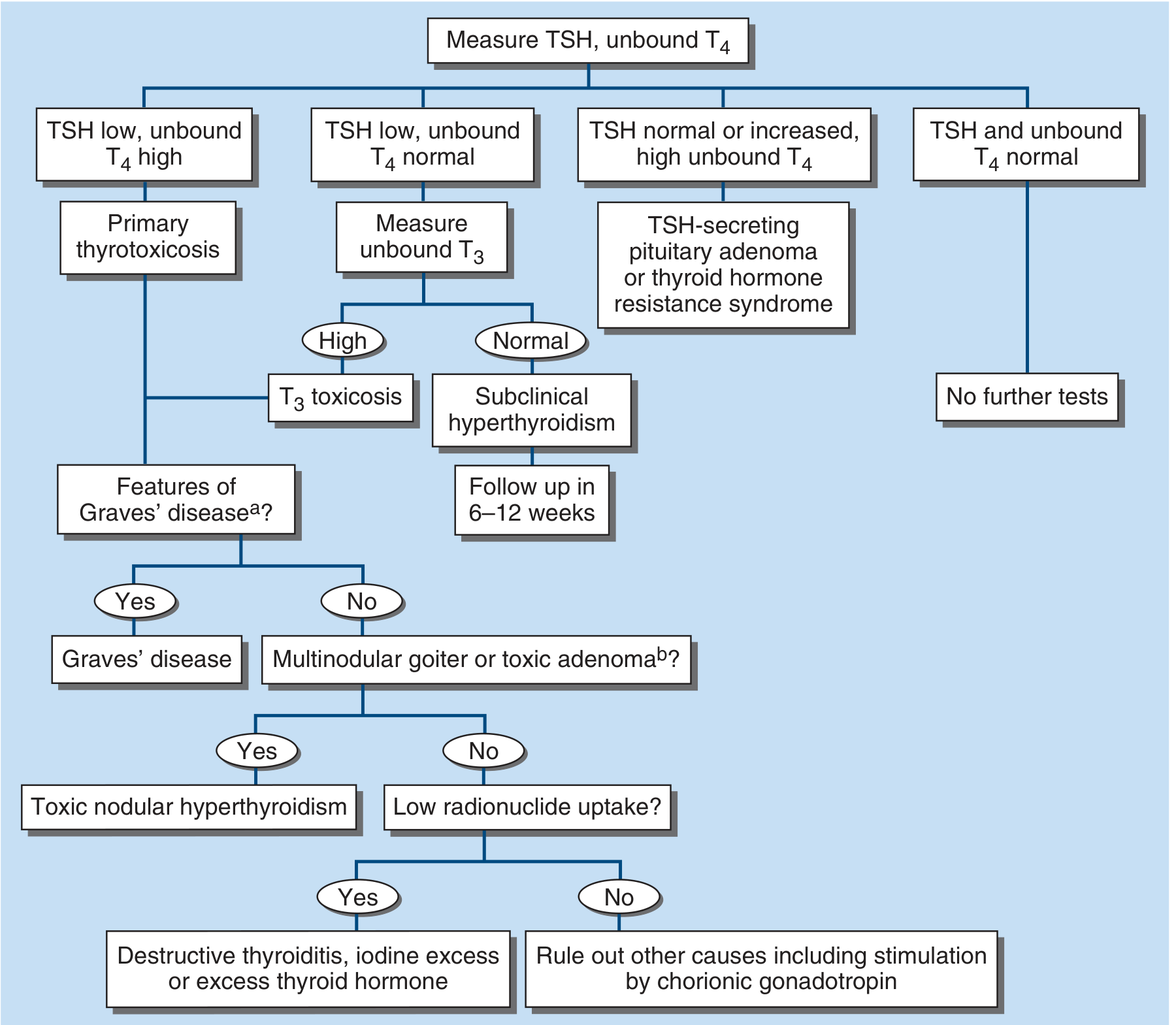
FIGURE 396-2: Evaluation of thyrotoxicosis. Start with TSH + unbound T4.
Key biochemical patterns:
- Primary hyperthyroidism: TSH suppressed (<0.1 mIU/L) + elevated free T4 and/or T3
- T3 toxicosis: TSH suppressed, T4 normal, only T3 elevated (2-5% of Graves'; more common with borderline iodine intake)
- Subclinical hyperthyroidism: TSH suppressed, T4 and T3 normal, no symptoms
- Secondary (pituitary): TSH NOT suppressed + elevated T4
For Graves' specifically: TRAb (TSH receptor antibody) measurement confirms diagnosis. Radionuclide scan shows diffuse HIGH uptake (distinguishes from destructive thyroiditis, which shows LOW uptake).
Treatment of Hyperthyroidism
Three main modalities:
1. Antithyroid Drugs (Thionamides)
- Methimazole (MMI) - preferred; blocks thyroid peroxidase, given once daily
- Propylthiouracil (PTU) - preferred in first trimester of pregnancy and thyroid storm (also blocks T4→T3 conversion); higher hepatotoxicity risk
- Side effects: agranulocytosis (0.2-0.5%) - must warn patients to stop drug and get WBC if fever/sore throat develops
2. Radioiodine (¹³¹I)
- First-line in most adults (US preference)
- Contraindicated in pregnancy
- Can worsen Graves' ophthalmopathy (especially in smokers)
- Most patients become hypothyroid within months-years and need LT4
3. Surgery (thyroidectomy)
- For large goiters, compressive symptoms, malignancy concern, pregnant patients failing PTU
- Patient must be rendered euthyroid pre-operatively
Adjunct therapy: Beta-blockers (propranolol, atenolol) for symptom control (tachycardia, tremor, anxiety) - do NOT lower thyroid hormone levels but block adrenergic effects.
Thyroid Storm (Thyrotoxic Crisis)
A life-threatening emergency triggered by infection, surgery, or trauma in an uncontrolled hyperthyroid patient. Mortality ~10-25%.
Management order is critical (from Rosen's Emergency Medicine):
| Step | Drug | Dose |
|---|---|---|
| 1. Beta-blocker FIRST | Propranolol 60-80 mg PO q4-6h | or IV: 0.5-1 mg slow push, then 1-2 mg q15 min |
| 2. Thionamide | PTU 500-1000 mg loading, then 250 mg q4h | or MMI 60-80 mg/day |
| 3. Iodine (≥1 hr AFTER thionamide) | Lugol's solution 5-7 drops TID | or SSKI 1-2 drops TID |
| 4. Corticosteroids | Hydrocortisone 300 mg IV, then 100 mg TID | Inhibits T4→T3 conversion, treats relative adrenal insufficiency |
| 5. Supportive | IV fluids, cooling, acetaminophen (NOT aspirin - displaces T4 from TBG) |
⚠️ Iodine MUST be given at least 1 hour AFTER the thionamide - if given first, it can precipitate thyroid storm by providing substrate for more hormone synthesis (Wolff-Chaikoff escape failure).
HYPOTHYROIDISM
Causes
Primary hypothyroidism (thyroid gland failure - most common, elevated TSH):
| Cause | Mechanism |
|---|---|
| Hashimoto's thyroiditis | Autoimmune destruction; most common cause in iodine-sufficient areas |
| Post-radioiodine | Gland destruction |
| Post-thyroidectomy | Surgical removal |
| Iodine deficiency | Most common cause worldwide |
| Drugs | Amiodarone, lithium, interferon, tyrosine kinase inhibitors |
| Subacute/postpartum thyroiditis | Transient hypothyroid phase |
| Congenital (cretinism) | Thyroid agenesis/dyshormonogenesis |
Central hypothyroidism (pituitary/hypothalamic failure - TSH low or inappropriately normal):
- Pituitary adenoma, Sheehan's syndrome, hypothalamic disease
Hashimoto's Thyroiditis - Deep Dive
The leading cause of hypothyroidism in the developed world. Autoimmune destruction mediated by T-lymphocytes. Antibodies present:
- Anti-TPO (anti-thyroid peroxidase) - most sensitive, >90% positive
- Anti-thyroglobulin (anti-Tg) - less specific
Patients may be euthyroid, hypothyroid, or occasionally transiently thyrotoxic ("Hashitoxicosis") early in disease. Goiter is common. Small increased risk of thyroid lymphoma.
Clinical Features of Hypothyroidism
| System | Symptoms | Signs |
|---|---|---|
| General | Fatigue, cold intolerance, weight gain, hoarse voice | Hypothermia, periorbital puffiness, slow speech |
| Cardiovascular | Dyspnea on exertion | Bradycardia, pericardial effusion, diastolic hypertension |
| Neuromuscular | Muscle cramps, weakness, depression, cognitive slowing | Delayed relaxation of deep tendon reflexes (hallmark), carpal tunnel |
| GI | Constipation | - |
| Skin/Hair | Dry skin, hair loss, brittle nails | Myxedema (non-pitting edema), coarse dry skin, loss of outer 1/3 eyebrow |
| Reproductive | Menorrhagia, infertility | Galactorrhea (elevated TRH also stimulates prolactin) |
"The signs and symptoms of hypothyroidism all lack sensitivity and specificity individually - TSH is the best screening test." - Symptom to Diagnosis, 4th Ed.
Diagnosis of Hypothyroidism
| Pattern | Interpretation |
|---|---|
| TSH elevated + free T4 low | Overt primary hypothyroidism |
| TSH elevated + free T4 normal | Subclinical hypothyroidism |
| TSH normal/low + free T4 low | Central (secondary/tertiary) hypothyroidism |
Anti-TPO antibodies confirm autoimmune etiology. For central hypothyroidism, TSH alone is unreliable - use free T4.
Treatment of Hypothyroidism
Levothyroxine (LT4) is the standard of care.
Dosing:
- Full replacement: 1.6 μg/kg/day (typically 100-150 μg/day) in patients without residual thyroid function
- Start low (50-100 μg/day) in patients <60 years without cardiac disease; go even lower (25 μg/day) in elderly or patients with known cardiac disease
- Goal: TSH in the normal range, ideally lower half of reference range
- Recheck TSH at 6-8 weeks after any dose change
- Follow-up TSH annually once stable
Take LT4 on an empty stomach (30-60 min before breakfast).
Drugs that impair LT4 absorption/increase requirements:
- Calcium supplements, ferrous sulfate, PPIs, cholestyramine, sucralfate
- Rifampicin, phenytoin, carbamazepine, amiodarone (increase T4 metabolism)
- Estrogens (increase TBG binding)
Subclinical hypothyroidism:
- Treat if: TSH >10 mIU/L, OR patient is pregnant/wants to conceive, OR symptomatic
- Observe if: TSH 4-10 mIU/L with no symptoms
Myxedema Coma
The extreme end of severe, long-standing hypothyroidism. A rare but highly lethal emergency (mortality 20-60% even with treatment).
Precipitants: Infection, cold exposure, drugs (opiates, sedatives), cardiac failure, non-compliance with LT4.
Features: Altered consciousness, hypothermia, bradycardia, hypoventilation, hyponatremia, hypoglycemia.
Treatment:
- IV levothyroxine (T4) 200-400 μg loading dose, then 50-100 μg/day
- Some add IV T3 (liothyronine) given slow T4-to-T3 peripheral conversion in critically ill
- IV hydrocortisone (concurrent adrenal insufficiency must be assumed and treated)
- Supportive: rewarming, ventilation, treat precipitant
- Begin IV T4 once clinical diagnosis is made - do NOT wait for lab confirmation (Plum & Posner's; Harrison's 22E)
Quick Comparison Table
| Feature | Hyperthyroidism | Hypothyroidism |
|---|---|---|
| TSH | Low (suppressed) | High (primary) |
| Free T4 | High | Low |
| Heart rate | Fast (tachycardia, AF) | Slow (bradycardia) |
| Weight | Loss (increased appetite) | Gain (decreased appetite) |
| Temperature | Heat intolerance, sweating | Cold intolerance |
| Reflexes | Hyperreflexia | Delayed relaxation (hung reflexes) |
| Bowel | Diarrhea, increased frequency | Constipation |
| Skin | Warm, moist, fine hair | Dry, coarse, hair loss, myxedema |
| Mood | Anxiety, irritability, insomnia | Depression, cognitive slowing |
| Metabolism | Hypermetabolic | Hypometabolic |
| Emergency | Thyroid storm | Myxedema coma |
| Main cause | Graves' disease | Hashimoto's thyroiditis |
| Treatment | Thionamides / ¹³¹I / surgery | Levothyroxine (LT4) |
Sources: Harrison's Principles of Internal Medicine 22E (2025), Rosen's Emergency Medicine, Sabiston Textbook of Surgery, Goodman & Gilman's Pharmacology, Scott-Brown's Otorhinolaryngology, Kaplan & Sadock's Psychiatry, Symptom to Diagnosis 4th Ed.
Diseases of pituitary gland
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
I now have comprehensive content from Harrison's 22E. Let me compile the full, well-structured answer.
Diseases of the Pituitary Gland
Anatomy & Hormones - Quick Review
The pituitary gland sits in the sella turcica at the base of the brain, connected to the hypothalamus via the pituitary stalk. It has two lobes:
| Lobe | Hormones |
|---|---|
| Anterior (adenohypophysis) | GH, TSH, ACTH, LH, FSH, Prolactin |
| Posterior (neurohypophysis) | ADH (Arginine Vasopressin - AVP), Oxytocin |
Pituitary diseases fall into three categories:
- Hypersecretion - from functioning adenomas
- Hyposecretion - hypopituitarism
- Mass effects - from tumors pressing on adjacent structures
PITUITARY ADENOMAS - Overview
Pituitary adenomas are the most common cause of pituitary dysfunction in adults. They account for ~15% of all intracranial neoplasms, with a population prevalence of ~80/100,000. At autopsy, up to one-quarter of all pituitary glands harbor an unsuspected microadenoma.
Classification by size:
- Microadenoma - <10 mm diameter; usually intrasellar, non-invasive
- Macroadenoma - ≥10 mm diameter; may invade parasellar structures
Classification by function:
- Functioning (secreting): Prolactinoma (most common), GH-secreting, ACTH-secreting, TSH-secreting, Gonadotropin-secreting
- Non-functioning (clinically silent): Cause mass effects only
MASS EFFECTS OF SELLAR LESIONS
Expanding pituitary tumors produce predictable effects based on the direction of growth:
| Structure Compressed | Clinical Effect |
|---|---|
| Optic chiasm | Bitemporal hemianopia (classic), superior field defect, blindness |
| Pituitary itself | Hypogonadism, hypothyroidism, hypoadrenalism, growth failure |
| Pituitary stalk | Stalk compression → hyperprolactinemia + loss of other hormones |
| Cavernous sinus | CN III, IV, VI palsies → diplopia, ptosis, ophthalmoplegia; facial numbness (V1, V2) |
| Hypothalamus | Temperature dysregulation, obesity, AVP deficiency, sleep disorders |
| Frontal lobe | Personality disorder, anosmia |
Headache is common even with small intrasellar tumors - due to stretching of the dural plate, and correlates poorly with adenoma size.
(Harrison's 22E, Table 392-1)
HYPERSECRETION SYNDROMES
1. PROLACTINOMA (Most Common Functioning Adenoma)
Epidemiology:
- Accounts for ~50% of all functioning pituitary tumors
- Prevalence: ~10/100,000 men; ~30/100,000 women
- Female:male ratio = 20:1 for microadenomas; ~1:1 for macroadenomas
Clinical Features:
| Women | Men |
|---|---|
| Amenorrhea (most common presentation) | Impotence, loss of libido |
| Galactorrhea | Infertility |
| Infertility | Gynecomastia |
| Oligomenorrhea | Often presents with mass effects (because male hypogonadism is subtle and delays diagnosis) |
Diagnosis:
- PRL level >200 μg/L = very likely prolactinoma
- PRL <100 μg/L = may be microadenoma OR stalk compression by another lesion (stalk effect)
- MRI pituitary (with gadolinium) in ALL patients with hyperprolactinemia
- Note: dopamine agonists can suppress PRL from ANY cause, not just prolactinoma
Causes of hyperprolactinemia (non-tumor):
Pregnancy, breastfeeding, hypothyroidism (elevated TRH stimulates PRL), renal failure, drugs (antipsychotics, metoclopramide, antidepressants, opioids), stalk compression by any lesion
Treatment:
- Dopamine agonists = first-line (suppress PRL synthesis + reduce tumor size)
- Cabergoline - long-acting (twice weekly dosing), higher D2 affinity, ~80-90% efficacy, preferred
- Bromocriptine - shorter-acting, preferred when pregnancy is desired
- Surgery (transsphenoidal) - for macroadenomas resistant or intolerant to dopamine agonists
- ~20% of patients (especially males) are dopamine agonist-resistant; surgery or radiation required
- Microadenomas that normalize on treatment may have dopamine agonist withdrawn after 2 years with close monitoring
2. ACROMEGALY / GIGANTISM (GH-Secreting Adenoma)
Definition:
- Gigantism = GH excess before epiphyseal closure (children) → tall stature
- Acromegaly = GH excess after epiphyseal closure (adults) → characteristic soft tissue and bony changes
Clinical Features of Acromegaly:
| System | Features |
|---|---|
| Face/Head | Frontal bossing, prognathism (jaw protrusion), macroglossia, widened teeth spacing, coarsening of facial features |
| Hands/Feet | Spade-like hands, broad feet, increased ring/shoe size |
| Soft tissue | Skin thickening, increased sweating (common complaint), oily skin |
| Cardiovascular | Cardiomegaly, hypertension, heart failure (leading cause of death) |
| Metabolic | Diabetes mellitus (GH is insulin-antagonist), dyslipidemia |
| Respiratory | Sleep apnea (soft tissue hypertrophy of upper airway) |
| Musculoskeletal | Arthropathy, carpal tunnel syndrome |
| Neurological | Headache, visual field defects (mass effect) |
| Colon | Increased risk of colon polyps and colon cancer (surveillance colonoscopy needed) |
Diagnosis:
- Screening: Random IGF-1 (elevated, age-matched)
- Confirmation: Oral glucose tolerance test (OGTT) - GH should suppress to <1 ng/mL after 75g glucose; failure to suppress confirms acromegaly
- MRI pituitary to localize the adenoma
Treatment:
The treatment algorithm from Harrison's 22E (FIGURE 392-3):
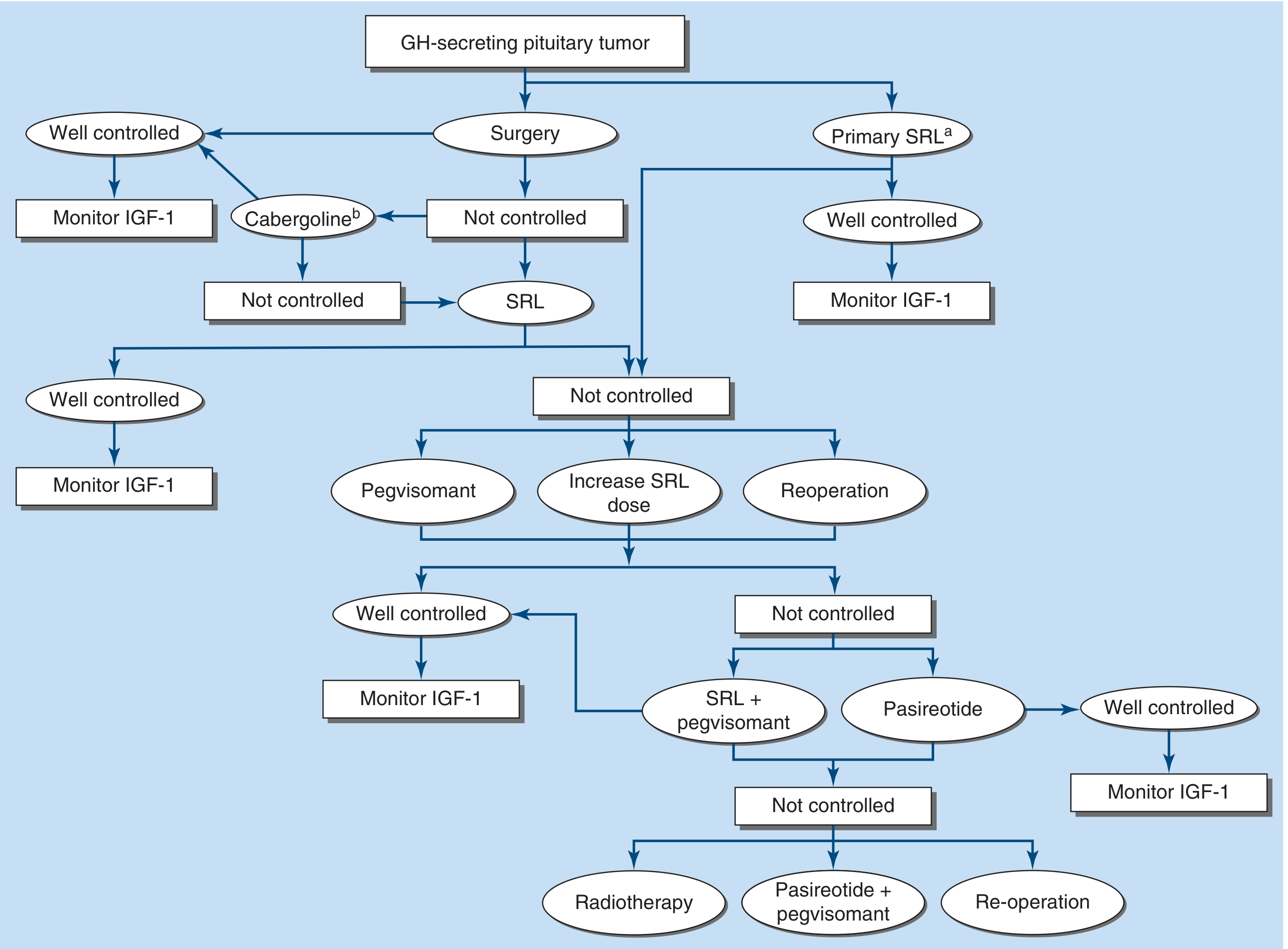
FIGURE 392-3: Management of GH-secreting pituitary tumor. Surgery is first-line; medical therapy follows for residual disease.
In sequence:
- Transsphenoidal surgery - first-line; cure rate ~80% for microadenomas, ~50% for macroadenomas
- Somatostatin receptor ligands (SRLs) - octreotide LAR or lanreotide (normalize GH/IGF-1 in ~50%); also used as primary therapy if surgery not feasible
- Pegvisomant - GH receptor antagonist; normalizes IGF-1 in >90%; used in SRL-resistant cases
- Cabergoline - for mild residual disease post-surgery
- Radiotherapy - last resort; slow effect over years, causes hypopituitarism
Biochemical cure criteria: GH nadir <1 ng/mL on OGTT AND normal age-matched IGF-1
3. CUSHING'S DISEASE (ACTH-Secreting Pituitary Adenoma)
Note: Cushing's DISEASE = pituitary ACTH-secreting adenoma specifically Cushing's SYNDROME = any cause of hypercortisolism (adrenal tumor, ectopic ACTH, exogenous steroids)
Epidemiology: Female predominance (F>M). Most tumors are microadenomas <5 mm - ~50% are not visible even on sensitive MRI.
Clinical Features of Cushing's Syndrome:
| Feature | Detail |
|---|---|
| Central obesity | Moon face, buffalo hump, truncal obesity with thin extremities |
| Skin | Violaceous striae (>1 cm wide), easy bruising, thin skin, poor wound healing, acne, hirsutism |
| Hypertension | Very common |
| Hyperglycemia / Diabetes | Cortisol is insulin-antagonist |
| Osteoporosis | Risk of fractures |
| Myopathy | Proximal muscle weakness (difficulty climbing stairs, rising from chair) |
| Psychiatric | Depression, psychosis, cognitive impairment |
| Hypogonadism | Menstrual irregularities, loss of libido |
Distinguishing Pituitary (Cushing's Disease) vs. Ectopic ACTH:
| Feature | Cushing's Disease | Ectopic ACTH |
|---|---|---|
| Sex | F > M | M > F |
| Onset | Slow, gradual | Rapid |
| Pigmentation | Absent | Present (very high ACTH) |
| Serum K+ <3.3 mmol/L | <10% | ~70% |
| Basal ACTH | Inappropriately high | Very high |
| High-dose dexamethasone suppression | Suppresses cortisol >50% | No suppression |
Diagnosis:
- 24-hour urinary free cortisol (UFC) - best screening test
- Overnight 1 mg dexamethasone suppression test - failure to suppress cortisol (<50 nmol/L) = positive
- Midnight serum/salivary cortisol - elevated (loss of normal diurnal rhythm)
- Inferior petrosal sinus sampling (IPSS) with CRH - gold standard to distinguish pituitary vs. ectopic ACTH when MRI is negative
Treatment:
- Transsphenoidal surgery - first-line (cure rate ~80% for microadenomas)
- Medical options (pre-op or when surgery fails): ketoconazole, metyrapone, osilodrostat, mifepristone, pasireotide (SST analogue effective specifically for Cushing's)
- Bilateral adrenalectomy - for refractory cases (risk of Nelson's syndrome: enlarging corticotrope adenoma post-adrenalectomy with extreme hyperpigmentation)
- Radiation - for recurrent/residual disease
4. TSH-SECRETING ADENOMA (Rare, ~1% of adenomas)
- Causes secondary hyperthyroidism with a NORMAL or ELEVATED TSH (paradoxically)
- Presents as hyperthyroidism + goiter + non-suppressed TSH
- Distinguish from thyroid hormone resistance syndrome
- Treatment: transsphenoidal surgery + octreotide (SST suppresses TSH)
5. NON-FUNCTIONING PITUITARY ADENOMAS
- No hormonal hypersecretion; often secrete gonadotropins (FSH, LH subunits) without clinical effect
- Present late with mass effects: visual field loss (bitemporal hemianopia), headache, hypopituitarism from compression
- Treatment: surgery (transsphenoidal) when causing symptoms; surveillance with serial MRI for small, asymptomatic lesions
HYPOPITUITARISM
Deficiency of one or more pituitary hormones. When all anterior pituitary hormones are lost = panhypopituitarism.
Causes
| Category | Examples |
|---|---|
| Tumors | Pituitary adenoma, craniopharyngioma, meningioma, glioma, metastases (breast, lung most common) |
| Vascular | Pituitary apoplexy (hemorrhage into adenoma), Sheehan's syndrome (postpartum necrosis) |
| Inflammatory/infiltrative | Lymphocytic hypophysitis, sarcoidosis, hemochromatosis, histiocytosis X (Langerhans cell histiocytosis) |
| Iatrogenic | Surgery, radiation therapy, immunotherapy (CTLA-4, PD-1 inhibitors) |
| Infection | TB, fungal (histoplasmosis), parasitic |
| Congenital | Kallmann syndrome (GnRH deficiency + anosmia), isolated GH deficiency |
| Traumatic | Head injury, skull base fractures, stalk compression |
Key principle (hormone loss order): GH → FSH/LH → TSH → ACTH (GH fails first, ACTH last) In children: growth retardation is the presenting feature In adults: hypogonadism (menstrual irregularities in women, impotence in men) is the earliest symptom
Sheehan's Syndrome
Postpartum pituitary necrosis due to severe hemorrhage/shock during delivery causing ischemia of the enlarged pituitary. Classic presentation: failure to lactate (PRL deficiency), failure of menses to resume, fatigue, cold intolerance (TSH/ACTH deficiency). Can present years later.
Pituitary Apoplexy
Sudden hemorrhage into a pituitary adenoma (or rarely normal gland). Emergency:
- Sudden severe headache, visual changes, ophthalmoplegia, meningism
- Cardiovascular collapse in severe cases
- Management: High-dose glucocorticoids (acute adrenal insufficiency); urgent surgical decompression if visual loss or altered consciousness
Clinical Features of Hypopituitarism by Hormone
| Hormone Lost | Clinical Features |
|---|---|
| GH | Children: short stature; Adults: central obesity, reduced muscle mass, fatigue, poor quality of life, dyslipidemia, cardiovascular risk |
| FSH/LH (Gonadotropins) | Women: amenorrhea, infertility, vaginal dryness; Men: impotence, infertility, loss of secondary sexual characteristics, testicular atrophy |
| TSH | Secondary hypothyroidism (same symptoms as primary, but TSH is low/normal) |
| ACTH | Secondary adrenal insufficiency: fatigue, weight loss, hypotension, hypoglycemia (no hyperkalemia since mineralocorticoids preserved; no hyperpigmentation since ACTH is low) |
| Prolactin | Failure of lactation postpartum |
Laboratory Diagnosis
- "Low trophic hormone + low target hormone" pattern
- e.g., Low free T4 + low/normal TSH = secondary hypothyroidism
- e.g., Low testosterone + low/normal LH = hypogonadotropic hypogonadism
- Insulin tolerance test (ITT): gold standard for GH and ACTH reserve (glucose <2.2 mmol/L with peak GH <3 ng/mL = GH deficiency; peak cortisol <500 nmol/L = ACTH deficiency)
Treatment of Hypopituitarism (Hormone Replacement)
| Hormone | Replacement |
|---|---|
| Cortisol (ACTH) | Hydrocortisone 15-25 mg/day (must replace FIRST before thyroid hormone) |
| Thyroid (TSH) | Levothyroxine (T4); monitor with free T4 levels, NOT TSH |
| Sex steroids (LH/FSH) | Testosterone (men); estrogen/progesterone (women); gonadotropins if fertility desired |
| Growth hormone | Recombinant human GH (rhGH) injections; indicated if symptomatic and biochemically confirmed |
| ADH (posterior) | Desmopressin (DDAVP) - nasal spray or oral |
⚠️ Always replace cortisol before thyroid hormone - giving T4 in unrecognized ACTH deficiency can precipitate adrenal crisis.
POSTERIOR PITUITARY DISORDERS
Arginine Vasopressin Deficiency (AVP-D) / Central Diabetes Insipidus
ADH (AVP) deficiency causes inability to concentrate urine.
- Causes: Pituitary/hypothalamic surgery, tumors (craniopharyngioma), trauma, sarcoidosis, histiocytosis X, metastases (posterior pituitary), idiopathic (~30%)
- Features: Polyuria (>3 L/day), polydipsia, dilute urine (low specific gravity <1.005), normal serum glucose
- Diagnosis: Water deprivation test - failure to concentrate urine, corrected by DDAVP (confirms central vs. nephrogenic DI)
- Treatment: Desmopressin (DDAVP) - synthetic AVP analogue, given intranasally, orally, or IV
SIADH (Syndrome of Inappropriate ADH Secretion)
- Excess AVP secretion: euvolemic hyponatremia, concentrated urine in the setting of dilute plasma
- Causes: CNS disease, ectopic (small cell lung cancer), drugs (carbamazepine, SSRIs, vincristine), postoperative state
- Treatment: fluid restriction; severe/symptomatic: hypertonic saline; tolvaptan (V2 receptor antagonist) for chronic SIADH
Summary Table: Pituitary Tumors at a Glance
| Tumor Type | Hormone | Prevalence | Classic Syndrome | Key Test | Treatment |
|---|---|---|---|---|---|
| Prolactinoma | PRL ↑ | Most common (~50%) | Amenorrhea-galactorrhea (women); impotence (men) | Serum PRL; MRI | Cabergoline (1st-line) |
| Somatotrope adenoma | GH ↑ | 2nd most common | Acromegaly (adults); gigantism (children) | IGF-1, OGTT GH | Surgery → SRLs |
| Corticotrope adenoma | ACTH ↑ | ~10-15% | Cushing's disease | 24h UFC, overnight DST | Surgery |
| TSH-oma | TSH ↑ | Rare (<1%) | Hyperthyroidism + non-suppressed TSH | TSH, alpha subunit | Surgery + octreotide |
| Non-functioning | None | ~30-40% | Mass effects only | MRI; visual fields | Surgery (if symptomatic) |
Sources: Harrison's Principles of Internal Medicine 22E (2025), Robbins & Kumar Basic Pathology, Scott-Brown's Otorhinolaryngology Vol 1, Neuroanatomy through Clinical Cases 3rd Ed., Goodman & Gilman's Pharmacology, Bradley and Daroff's Neurology in Clinical Practice
Explain in simple about adh hormone physiology
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
I now have excellent, comprehensive material. Let me present the full simple-language explanation.
ADH (Antidiuretic Hormone) Physiology - Simple Explanation
What is ADH?
ADH stands for Anti-Diuretic Hormone (also called Vasopressin or Arginine Vasopressin / AVP).
- "Anti" = against
- "Diuretic" = making urine
- So ADH literally means: "hormone that prevents excess urine production"
Its main job is to keep water inside your body by telling the kidneys to hold onto it rather than pee it out.
Where is it Made and Stored?
Hypothalamus (makes it)
↓
Travels down the pituitary stalk
↓
Posterior Pituitary (stores & releases it)
ADH is synthesized in two clusters of nerve cell bodies in the hypothalamus:
- Supraoptic nucleus
- Paraventricular nucleus
These neurons send their axons all the way down to the posterior pituitary gland, where ADH is stored in granules, ready to be released into the bloodstream on demand.
Think of it like a water factory (hypothalamus) connected to a warehouse (posterior pituitary) via a delivery pipe (pituitary stalk).
The Big Idea: Why Does the Body Need ADH?
Your blood needs to maintain the right concentration at all times. This concentration is measured as osmolality (normal = 280-295 mOsm/kg).
- If you get dehydrated (not enough water) → blood becomes too concentrated (high osmolality) → DANGER
- If you drink too much water → blood becomes too dilute (low osmolality) → also DANGER
ADH is the solution. It acts like a dimmer switch for how much water the kidneys hold onto.
What Triggers ADH Release? (Stimuli)
1. High Blood Osmolality (Most Sensitive Trigger)
When blood becomes too concentrated (e.g., you're dehydrated, sweating, not drinking enough):
- Special sensors called osmoreceptors in the hypothalamus detect this
- Even a 1-2% rise in blood osmolality is enough to trigger ADH release
- ADH is released → kidneys hold onto water → urine becomes dark and concentrated → osmolality returns to normal
2. Low Blood Volume / Low Blood Pressure
When you lose blood, vomit severely, or are in shock:
- Baroreceptors (pressure sensors) in the carotid artery and aorta detect the drop
- They signal the hypothalamus to release ADH
- But this requires a much bigger stimulus - a 5-10% drop in blood volume is needed (less sensitive than osmolality)
3. Other Stimulants
- Nausea (very powerful ADH trigger - this is why vomiting can cause water retention)
- Pain
- Stress
- Nicotine
- Hypoglycemia (low blood sugar)
- Some drugs (opioids, carbamazepine, cyclophosphamide)
What SUPPRESSES ADH?
- Drinking lots of water (low osmolality)
- Alcohol (alcohol blocks ADH → you urinate more after drinking → dehydration and hangover)
- Atrial natriuretic peptide (ANP)
How Does ADH Work on the Kidney?
This is the heart of ADH physiology - a beautiful 4-step mechanism:
ADH released into blood
↓
Travels to kidney collecting duct
↓
Binds to V2 receptors on collecting duct cells
↓
Activates adenylate cyclase → ↑ cAMP
↓
cAMP moves water channels (Aquaporin-2) to cell surface
↓
Water flows from urine back into blood
↓
Urine becomes concentrated, less volume produced
The key players:
| Player | Role |
|---|---|
| V2 receptor | ADH's docking station on kidney cells |
| cAMP | Messenger molecule inside the cell |
| Aquaporin-2 (AQP2) | Water channel / "water door" in the cell membrane |
| Aquaporin-3 & 4 | Water channels on the other side of the cell (exit route into blood) |
Simple analogy:
Imagine the collecting duct is a pipe with lots of tiny "water doors" (aquaporins) stored in a closet. Normally the doors are locked away. When ADH arrives, it sends a signal that opens the closet and installs water doors into the pipe wall. Now water can flow from the pipe (urine) back into the body (blood). When ADH is gone, the doors get taken off the wall and put back in the closet.
The ADH-Osmolality Relationship
This is a straight-line (linear) relationship:
| Blood Osmolality | ADH Level | Urine Output |
|---|---|---|
| <284 mOsm/kg | Suppressed (near zero) | Large volume, very dilute (up to 18-24 L/day) |
| 284-295 mOsm/kg | 0.5-2 pg/mL (basal) | Normal (1-3 L/day) |
| >295 mOsm/kg | Maximally elevated | Small volume, very concentrated (max ~1200 mOsm/kg) |
Without ANY ADH: urine output can reach 800-1000 mL per hour! (18-24 L/day) - this is what happens in untreated Diabetes Insipidus.
The Role of THIRST
Thirst and ADH work together but are separate systems:
Dehydration → Blood osmolality rises
↙ ↘
ADH released Thirst activated
(kidney saves water) (you drink water)
Thirst kicks in at a slightly higher osmolality (~5-10 mOsm/kg above the ADH threshold). This makes sense:
- ADH is the first line of defense - tries to fix the problem quietly (retaining water)
- Thirst is the backup - makes you actively seek water when dehydration is more severe
Under normal daily life, ADH is more important than thirst for maintaining water balance. Thirst becomes critical only in severe dehydration.
ADH's Other Receptors (Beyond the Kidney)
ADH acts on 3 types of receptors:
| Receptor | Location | Effect |
|---|---|---|
| V1a | Blood vessel smooth muscle | Vasoconstriction → raises blood pressure (that's why it's also called "Vasopressin") |
| V1b | Anterior pituitary | Stimulates ACTH release |
| V2 | Kidney collecting duct | Water reabsorption via AQP2 (main function) |
At normal blood levels, ADH mainly works on V2 (kidney). It only causes significant vasoconstriction at HIGH (pharmacologic) doses - which is why vasopressin is used as a drug in septic shock.
ADH and Clotting Factor VIII
ADH also has a bonus function: it stimulates release of von Willebrand factor and clotting factor VIII from endothelial cells. This is why the synthetic form (DDAVP/desmopressin) is used to treat bleeding disorders like mild haemophilia A and von Willebrand disease.
What Happens When ADH Goes Wrong?
Too LITTLE ADH (or kidney ignores it) = Diabetes Insipidus (DI)
| Type | Problem | Result |
|---|---|---|
| Central DI | Posterior pituitary doesn't make/release enough ADH | Massive dilute urine (polyuria), thirst, dehydration |
| Nephrogenic DI | Kidney V2 receptors don't respond to ADH | Same symptoms, but ADH levels are high |
Common causes of Central DI: Brain surgery, head trauma, tumors (craniopharyngioma), Sheehan's syndrome, sarcoidosis, idiopathic (~30%)
Treatment: DDAVP (desmopressin) - a synthetic ADH analogue that preferentially binds V2
Too MUCH ADH = SIADH (Syndrome of Inappropriate ADH)
The body retains too much water → blood becomes too dilute → hyponatremia (low sodium).
Causes: Small cell lung cancer (ectopic ADH), CNS disease (meningitis, stroke), drugs (SSRIs, carbamazepine, cyclophosphamide), postoperative state, hypothyroidism, Addison's disease.
Classic lab pattern:
- Low serum sodium (<135 mmol/L)
- Low plasma osmolality
- Urine osmolality inappropriately HIGH (>100 mOsm/kg) - kidney still concentrating because of excess ADH
- Urine sodium >20 mmol/L
Treatment: Fluid restriction; hypertonic saline for severe/symptomatic hyponatremia; Tolvaptan (V2 receptor blocker) for chronic SIADH.
Quick Summary Diagram
DEHYDRATION / HIGH OSMOLALITY / LOW BP
↓
Hypothalamus osmoreceptors / baroreceptors
↓
ADH released from posterior pituitary
↓
ADH → V2 receptor on kidney collecting duct
↓
↑ cAMP → AQP2 water channels inserted
↓
Water saved from urine → back into blood
↓
Blood osmolality normalizes, urine becomes concentrated
WELL-HYDRATED / LOW OSMOLALITY
↓
ADH suppressed
↓
AQP2 channels removed from membrane
↓
Water passes out as dilute urine
Sources: Goldman-Cecil Medicine, Henry's Clinical Diagnosis and Management by Laboratory Methods, Comprehensive Clinical Nephrology 7th Ed., Tietz Textbook of Laboratory Medicine 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.