📚 Pharm D 1st Year — Biochemistry Complete Notes
═══════════════════════════════════════
LONG ANSWER QUESTIONS (15 Marks Each)
═══════════════════════════════════════
LONG ANSWER 1 — Urea Cycle: Detailed Note with Reactions & Metabolic Disorders
🔑 Introduction
Urea (H₂N–CO–NH₂) is the major disposal form of amino groups derived from amino acid catabolism. It accounts for ~90% of nitrogen-containing components of urine. Urea synthesis occurs exclusively in hepatocytes (liver cells). The cycle was first described in 1932 by Hans Krebs and Kurt Henseleit (originally called the Krebs–Henseleit cycle).
Key fact: Normal blood urea nitrogen (BUN) = 7–20 mg/dL; plasma ammonia (NH₄⁺) = 30–60 µmol/L
🔬 Source of Nitrogen for Urea
| Nitrogen Atom | Source |
|---|
| 1st Nitrogen | Free NH₃ (from glutamate via GDH — oxidative deamination) |
| 2nd Nitrogen | Aspartate (via transamination) |
| Carbon & Oxygen | CO₂ (as HCO₃⁻) |
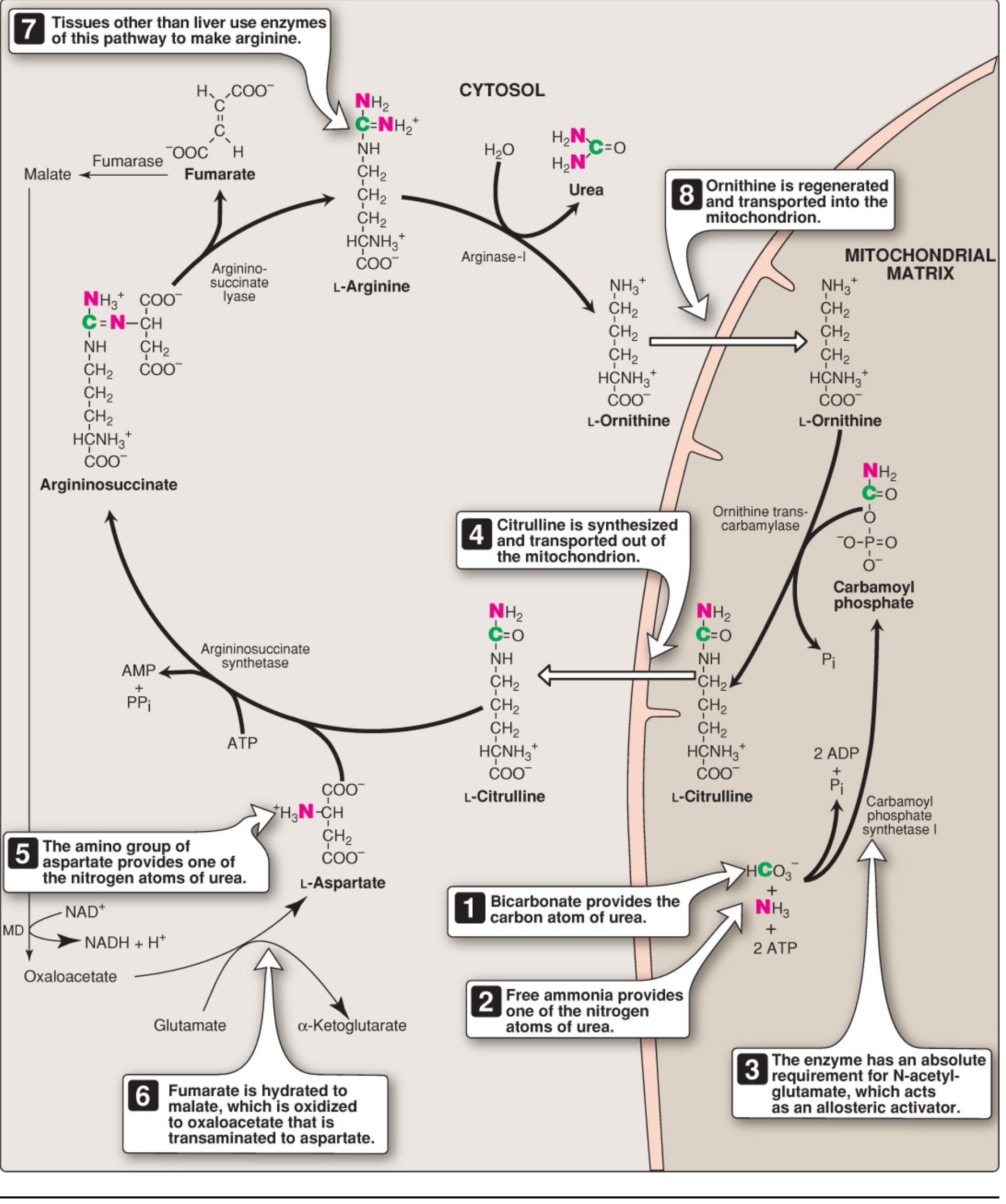
⚙️ Reactions of the Urea Cycle
The urea cycle has 5 major reactions. Steps 1–2 occur in the mitochondrial matrix; Steps 3–5 occur in the cytosol.
FLOWCHART — Urea Cycle
MITOCHONDRIA
───────────────────────────────────────────────────────
NH₄⁺ + HCO₃⁻ + 2ATP
↓ [CPS-I — activated by N-acetylglutamate (NAG)]
① CARBAMOYL PHOSPHATE (high-energy compound)
↓ + Ornithine
↓ [Ornithine Transcarbamylase (OTC)]
② CITRULLINE + Pᵢ
↓ (transported to cytosol via antiporter)
───────────────────────────────────────────────────────
CYTOSOL
───────────────────────────────────────────────────────
Citrulline + Aspartate + ATP
↓ [Argininosuccinate Synthetase] → AMP + PPᵢ
③ ARGININOSUCCINATE
↓ [Argininosuccinate Lyase]
④ ARGININE + Fumarate
↓ [Arginase-I (liver only)] + H₂O
⑤ UREA + ORNITHINE ←────────────────────────────────┐
│
Ornithine transported back to mitochondria ──┘
📋 Step-by-Step Reactions (Tabular)
| Step | Reaction | Enzyme | Location | Energy |
|---|
| 1 | NH₄⁺ + HCO₃⁻ + 2ATP → Carbamoyl phosphate | CPS-I (activated by NAG) | Mitochondria | 2 ATP consumed |
| 2 | Carbamoyl-P + Ornithine → Citrulline + Pᵢ | OTC (Ornithine Transcarbamylase) | Mitochondria | — |
| 3 | Citrulline + Aspartate + ATP → Argininosuccinate | Argininosuccinate Synthetase | Cytosol | 1 ATP → AMP + PPᵢ |
| 4 | Argininosuccinate → Arginine + Fumarate | Argininosuccinate Lyase | Cytosol | — |
| 5 | Arginine + H₂O → Urea + Ornithine | Arginase-I | Cytosol (liver) | — |
Overall Stoichiometry:
NH₄⁺ + HCO₃⁻ + Aspartate + 3 ATP + H₂O
→ Urea + Fumarate + 2 ADP + AMP + 2 Pᵢ + PPᵢ
4 high-energy phosphate bonds are consumed per mole of urea. The cycle is irreversible (large negative ΔG).
🧪 Regulation of the Urea Cycle
| Regulator | Effect | Mechanism |
|---|
| N-Acetylglutamate (NAG) | Activates CPS-I (rate-limiting enzyme) | Allosteric activator — increases affinity of CPS-I for ATP |
| Arginine | Activates NAGS | Stimulates NAG synthesis → more CPS-I activation |
| Substrate availability | Short-term regulation | More amino acid intake → more NH₃ → more urea |
| Enzyme induction | Long-term regulation | High-protein diet upregulates urea cycle enzymes |
⚠️ Major Metabolic Disorders of the Urea Cycle
When any enzyme of the urea cycle is deficient → Hyperammonemia (ammonia accumulates) → Toxic to CNS.
FLOWCHART — Urea Cycle Disorders
Step 1 → CPS-I deficiency → ↑ NH₄⁺ (no carbamoyl phosphate formed)
Step 2 → OTC deficiency → ↑ NH₄⁺ + ↑ Orotic acid in urine (most common; X-linked)
Step 3 → AS deficiency → Citrullinemia (↑ citrulline in blood/urine)
Step 4 → AL deficiency → Argininosuccinic acidemia (↑ argininosuccinate)
Step 5 → Arginase deficiency → Argininemia (↑ arginine in blood)
| Disorder | Deficient Enzyme | Key Feature | Inheritance |
|---|
| CPS-I deficiency | Carbamoyl Phosphate Synthetase-I | Severe hyperammonemia; no orotic acid | AR |
| OTC deficiency | Ornithine Transcarbamylase | Most common; ↑ orotic acid in urine | X-linked recessive |
| Citrullinemia | Argininosuccinate Synthetase | ↑ Citrulline in blood & urine | AR |
| Argininosuccinic aciduria | Argininosuccinate Lyase | ↑ Argininosuccinate; brittle hair (trichorrhexis nodosa) | AR |
| Hyperargininemia | Arginase-I | ↑ Arginine; spastic diplegia, seizures | AR |
AR = Autosomal Recessive
Common Clinical Features of ALL Urea Cycle Disorders:
- Neonatal vomiting, lethargy
- Hyperammonemia → Cerebral edema → Coma → Death
- Protein aversion (older children)
- Treatment: Low-protein diet + Sodium benzoate/phenylbutyrate (alternative nitrogen excretion pathways)
Other causes of Hyperammonemia:
- Acquired (Liver disease): Viral hepatitis, alcoholic cirrhosis → shunting of portal blood → urea cycle bypassed
- Liver failure → ammonia cannot be converted to urea → encephalopathy
LONG ANSWER 2 — Transamination Reaction in Catabolism of Amino Acids
🔑 Introduction
Transamination is the most important initial step in the catabolism of most amino acids. In this reaction, the α-amino group of an amino acid is transferred to an α-keto acid, producing a new amino acid and a new keto acid.
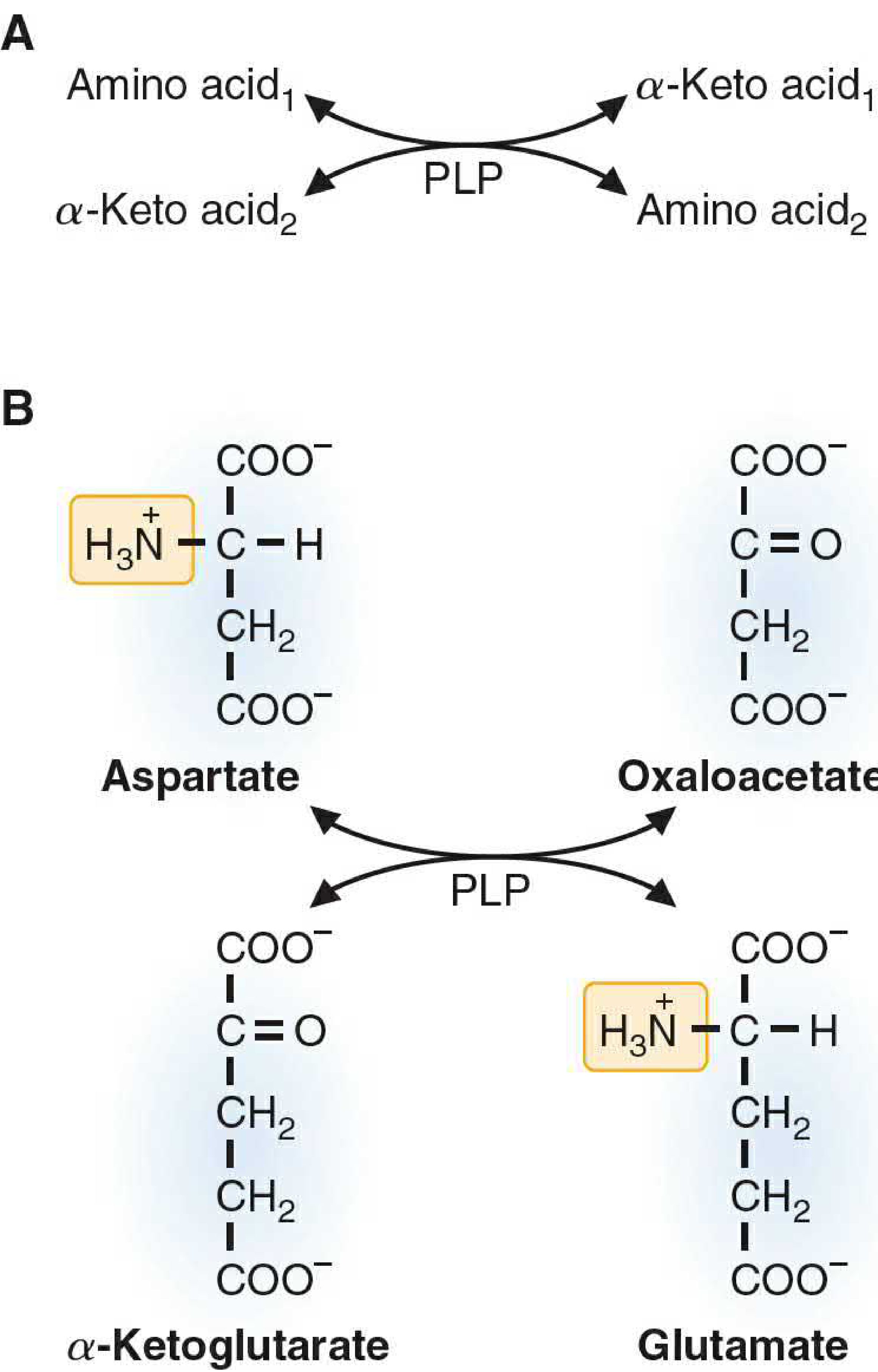
⚙️ General Reaction
Amino Acid₁ + α-Keto acid₂
↕ [Transaminase / Aminotransferase + PLP]
α-Keto acid₁ + Amino Acid₂
The most common pair:
Amino acid + α-Ketoglutarate ⇌ α-Keto acid + Glutamate
[Transaminase + PLP]
Example — Aspartate Transaminase (AST):
Aspartate + α-Ketoglutarate ⇌ Oxaloacetate + Glutamate
Example — Alanine Transaminase (ALT):
Alanine + α-Ketoglutarate ⇌ Pyruvate + Glutamate
🔬 Key Features
| Feature | Detail |
|---|
| Enzymes | Transaminases / Aminotransferases |
| Coenzyme required | Pyridoxal Phosphate (PLP) — derived from Vitamin B₆ |
| Reversibility | Reactions are readily reversible |
| Exceptions | Lysine and Threonine do NOT undergo transamination |
| Central acceptor | α-Ketoglutarate is almost always one of the pairs (produces glutamate) |
🔄 Mechanism (Step-by-Step)
Phase 1 — Amino group transfer TO enzyme (PLP → PMP):
Amino Acid + Enzyme-PLP → α-Keto Acid + Enzyme-PMP
Phase 2 — Amino group transfer FROM enzyme (PMP → PLP):
Enzyme-PMP + α-Ketoglutarate → Glutamate + Enzyme-PLP
PLP = Pyridoxal Phosphate (Schiff base with enzyme lysine)
PMP = Pyridoxamine Phosphate
🔗 Role of Transamination in Amino Acid Catabolism (FLOWCHART)
Dietary Amino Acids (20 types)
↓ Transamination (collects α-amino groups)
↓ All nitrogen funneled → GLUTAMATE
↓
Glutamate Dehydrogenase (GDH)
↓ Oxidative Deamination
α-Ketoglutarate + NH₄⁺
↓
NH₄⁺ enters → UREA CYCLE → Urea (excreted)
α-Keto acids → TCA cycle / gluconeogenesis / ketogenesis
🔗 Link with Other Pathways
| α-Keto Acid Produced | Metabolic Fate |
|---|
| Pyruvate (from Alanine) | Gluconeogenesis, TCA cycle |
| Oxaloacetate (from Aspartate) | TCA cycle, gluconeogenesis |
| α-Ketoglutarate (from Glutamate) | TCA cycle |
| Acetoacetate (from Leucine/Lys) | Ketogenesis |
🏥 Clinical Importance of Transamination
- Basis of diagnostic importance of transaminases (see Long Answer 4)
- Reversible reactions allow biosynthesis of non-essential amino acids
- Central to the glucose–alanine cycle (muscle → liver nitrogen transport)
LONG ANSWER 3 — Bile Pigments: Types, Metabolism & Associated Diseases
🔑 What Are Bile Pigments?
Bile pigments are colored degradation products of heme (the iron-containing prosthetic group of hemoglobin). They are excreted in bile. The principal bile pigments are:
- Bilirubin (yellow-orange) — major bile pigment
- Biliverdin (green) — intermediate
- Urobilinogen (colorless) — intestinal reduction product
- Urobilin / Stercobilin (yellow/brown) — oxidized urobilinogen
⚙️ Metabolism of Bilirubin (Step-by-Step Flowchart)
┌─────────────────────────────────────────────────────────┐
│ RBC breakdown (in spleen/liver/bone marrow) │
│ Hemoglobin → Heme + Globin │
│ Heme + O₂ → [Heme Oxygenase] → Biliverdin + CO + Fe²⁺ │
│ Biliverdin → [Biliverdin Reductase] → BILIRUBIN │
│ (Unconjugated / Indirect / Free) │
└──────────────────┬──────────────────────────────────────┘
│ Transported bound to ALBUMIN in blood
▼
┌─────────────────────────────────────────────────────────┐
│ LIVER (Hepatocytes) │
│ Bilirubin → [UDP-Glucuronyl Transferase] │
│ → Bilirubin Diglucuronide │
│ (Conjugated / Direct / Water-soluble) │
└──────────────────┬──────────────────────────────────────┘
│ Excreted into bile → intestine
▼
┌─────────────────────────────────────────────────────────┐
│ INTESTINE │
│ Bilirubin glucuronide → [Intestinal bacteria] │
│ → Urobilinogen (colorless) │
│ ├─ 20% reabsorbed → portal blood → liver │
│ │ (small amount → kidney → URINE │
│ │ as Urobilin = yellow color of urine) │
│ └─ 80% oxidized → Stercobilin │
│ (brown color of feces) │
└─────────────────────────────────────────────────────────┘
📋 Types of Bilirubin
| Property | Unconjugated (Indirect) | Conjugated (Direct) |
|---|
| Solubility | Water-INSOLUBLE | Water-SOLUBLE |
| Transport | Bound to albumin | Free in plasma |
| Van den Bergh test | Indirect reaction | Direct reaction |
| Crosses BBB | Yes (toxic to brain) | No |
| Present in urine | No (not filtered) | Yes (bilirubinuria) |
| Normal plasma level | ~0.1–0.8 mg/dL | <0.2 mg/dL |
Total normal serum bilirubin: 0.3–1.0 mg/dL
Jaundice appears when bilirubin >1.5–2 mg/dL (visually detectable ~3× normal)
🏥 Diseases Associated with Bile Pigment Metabolism
| Disease | Type | Defect | Bilirubin Type ↑ |
|---|
| Hemolytic Jaundice | Pre-hepatic | Excessive RBC destruction → excess bilirubin formation | Unconjugated ↑↑ |
| Hepatocellular Jaundice | Hepatic | Liver cell damage (hepatitis, cirrhosis) → impaired uptake + conjugation + excretion | Both types ↑ |
| Obstructive Jaundice | Post-hepatic | Bile duct obstruction (gallstone, cancer) → conjugated bilirubin regurgitated | Conjugated ↑↑ |
| Neonatal Jaundice | Physiological | Immature UDP-glucuronyl transferase | Unconjugated ↑ |
| Crigler-Najjar Syndrome | Hereditary | Absent (Type I) or deficient (Type II) UDP-glucuronyl transferase | Unconjugated ↑↑↑ |
| Gilbert Syndrome | Hereditary | Reduced UDP-glucuronyl transferase (~30% activity) | Mild unconjugated ↑ |
| Dubin-Johnson Syndrome | Hereditary | Defect in MRP2 (cannot excrete conjugated bilirubin into bile) | Conjugated ↑ |
| Rotor Syndrome | Hereditary | Defect in hepatic uptake and storage | Conjugated ↑ |
| Kernicterus | Complication | Unconjugated bilirubin crosses BBB → brain damage in neonates | Unconjugated ↑↑↑ |
LONG ANSWER 4 — Transaminases: Definition & Diagnostic Importance
🔑 Definition
Transaminases (also called Aminotransferases) are enzymes that catalyze transamination reactions — the transfer of an amino group (–NH₂) from an amino acid to an α-keto acid. All require pyridoxal phosphate (PLP) as coenzyme.
🔬 Clinically Important Transaminases
| Enzyme | Full Name | Old Name | Primary Location |
|---|
| AST | Aspartate Aminotransferase | SGOT (Serum Glutamate Oxaloacetate Transaminase) | Liver, heart, muscle, RBCs |
| ALT | Alanine Aminotransferase | SGPT (Serum Glutamate Pyruvate Transaminase) | Liver (most specific) |
Reactions:
AST: Aspartate + α-Ketoglutarate ⇌ Oxaloacetate + Glutamate
ALT: Alanine + α-Ketoglutarate ⇌ Pyruvate + Glutamate
Normal Reference Values:
| Enzyme | Normal Range |
|---|
| AST | Up to ~40 IU/L |
| ALT | Up to ~40 IU/L |
Blood half-life: AST = 17 h; ALT = 47 h; Mitochondrial AST = 87 h
🏥 Diagnostic Importance of Transaminases
FLOWCHART — Disease Diagnosis Using AST & ALT
Serum Transaminases Elevated
/ \
ALT > AST AST >> ALT (ratio >2:1)
| |
Acute Liver Disease Alcoholic Hepatitis
(Viral Hepatitis, Drugs) (DeRitis Ratio = AST/ALT ≥3:1)
| Condition | AST | ALT | AST/ALT Ratio | Notes |
|---|
| Viral Hepatitis (Acute) | ↑↑↑ (500–5000 IU/L) | ↑↑↑↑ (higher than AST) | <1 | ALT more specific for liver |
| Alcoholic Hepatitis | ↑↑ | ↑ (lower) | ≥2–3:1 (DeRitis ratio) | Mitochondrial AST released by alcohol |
| Myocardial Infarction (MI) | ↑↑ | Normal/mildly ↑ | >1 | AST from heart muscle (troponin now preferred) |
| Liver Cirrhosis | ↑ | ↑ | Variable | Reduced synthesis of both |
| Obstructive Jaundice | Mildly ↑ | Mildly ↑ | ~1 | ALP more prominently elevated |
| Drug-induced hepatotoxicity | ↑↑ | ↑↑↑ | <1 | Paracetamol overdose |
| Muscle Disease (Myopathy) | ↑↑ | Normal | >3 | AST from muscle cells |
Key Points for Exam:
- ALT is LIVER-SPECIFIC — predominantly found in liver cytoplasm; most sensitive marker of hepatocellular damage
- AST is LESS SPECIFIC — found in liver, heart, skeletal muscle, kidneys, RBCs
- DeRitis Ratio (AST/ALT):
- >2 → Alcoholic liver disease
- <1 → Viral hepatitis / non-alcoholic fatty liver
- Vitamin B₆ deficiency can give falsely LOW transaminase levels (PLP is required for enzyme activity — especially important in alcoholics)
- Both enzymes are used to monitor liver function, assess severity of hepatitis, guide treatment decisions, and detect drug toxicity
LONG ANSWER 5 — Porphyrins: Types and Clinical Importance of Porphyria
🔑 What Are Porphyrins?
Porphyrins are cyclic compounds formed by four pyrrole rings linked by methene bridges (–CH=). They have a unique ability to bind metal ions:
- Fe²⁺ → Heme (in hemoglobin, myoglobin, cytochromes)
- Mg²⁺ → Chlorophyll (plants)
- Co²⁺ → Vitamin B₁₂
Porphyrins are the backbone of heme, which is critical for oxygen transport and electron transfer.
⚙️ Heme Synthesis (Simplified)
MITOCHONDRIA:
Succinyl CoA + Glycine → [ALA Synthase + PLP] → δ-ALA (aminolevulinic acid)
CYTOSOL:
2× δ-ALA → [ALA Dehydratase] → Porphobilinogen (PBG) — 1 pyrrole ring
4× PBG → [Hydroxymethylbilane Synthase] → Hydroxymethylbilane
→ Uroporphyrinogen III → Coproporphyrinogen III
MITOCHONDRIA:
Coproporphyrinogen III → Protoporphyrin IX
Protoporphyrin IX + Fe²⁺ → [Ferrochelatase] → HEME
🔬 Types of Porphyria
Porphyrias are a group of diseases caused by enzyme defects in the heme biosynthetic pathway → accumulation of porphyrins or their precursors (ALA, PBG).
Classification:
PORPHYRIAS
│
├── HEPATIC (liver is primary site of overproduction)
│ ├── Acute Intermittent Porphyria (AIP) ← most common acute
│ ├── Porphyria Cutanea Tarda (PCT) ← most common overall
│ ├── Hereditary Coproporphyria (HCP)
│ └── Variegate Porphyria (VP)
│
└── ERYTHROPOIETIC (bone marrow/RBCs)
├── Congenital Erythropoietic Porphyria (CEP) ← Günther disease
└── Erythropoietic Protoporphyria (EPP)
📋 Major Types — Detailed Table
| Type | Deficient Enzyme | Precursor/Porphyrin Accumulated | Key Clinical Features | Inheritance |
|---|
| AIP (Acute Intermittent Porphyria) | Hydroxymethylbilane synthase (HMB synthase) / PBG deaminase | ALA, PBG (urine turns red/dark on standing) | Abdominal pain, neuropsychiatric symptoms, no photosensitivity | AD |
| Porphyria Cutanea Tarda (PCT) | Uroporphyrinogen decarboxylase | Uroporphyrin I & III | Photosensitivity, skin blistering, fragile skin, dark urine | Acquired (most common) |
| Congenital Erythropoietic Porphyria (CEP) | Uroporphyrinogen III synthase | Uroporphyrin I | Severe photosensitivity, hemolytic anemia, pink urine, red-stained teeth (erythrodontia) | AR |
| Erythropoietic Protoporphyria (EPP) | Ferrochelatase | Protoporphyrin IX | Photosensitivity (burning, itching), liver damage | AD |
| Hereditary Coproporphyria (HCP) | Coproporphyrinogen oxidase | Coproporphyrin III | Abdominal pain + photosensitivity | AD |
| Variegate Porphyria (VP) | Protoporphyrinogen oxidase | Porphyrins + ALA/PBG | Abdominal pain + photosensitivity; "South African type" | AD |
AD = Autosomal Dominant; AR = Autosomal Recessive
⚠️ Clinical Importance of Porphyria
Two Main Clinical Manifestations:
ACUTE ATTACKS (Neuropsychiatric) CUTANEOUS (Photosensitivity)
—————————————————————— ——————————————————————————
Caused by ALA + PBG accumulation Caused by porphyrin accumulation
(toxic to nervous system) (porphyrins absorb light → O₂ radicals)
Symptoms: Symptoms:
• Severe colicky abdominal pain • Blistering, erosions on sun-exposed skin
• Nausea, vomiting • Hyperpigmentation
• Motor neuropathy (weakness/paralysis) • Fragile skin
• Neuropsychiatric symptoms • Hypertrichosis
• Tachycardia, hypertension • Red/pink urine or teeth
Precipitating Factors for Acute Attacks:
- Drugs (barbiturates, sulfonamides, alcohol, estrogens) — induce ALA synthase
- Fasting/starvation
- Hormonal changes (menstruation, pregnancy)
- Infections
Diagnosis:
- ↑ Urinary ALA and PBG (Watson-Schwartz test — positive in AIP)
- Urine turns port-wine red on standing (oxidation of PBG)
- Stool porphyrins
- Fluorescence of porphyrins under UV light (Wood's lamp)
Treatment:
- Acute attack: IV Glucose (suppresses ALA synthase) + IV Hemin (negative feedback on ALA synthase)
- Avoid precipitating drugs
- Sunscreen and sun protection (for cutaneous types)
═══════════════════════════════════
SHORT ANSWER QUESTIONS (5 Marks)
═══════════════════════════════════
SHORT ANSWER 1 — Transamination Reaction in Catabolism of Amino Acids
Transamination = Transfer of the α-amino group from an amino acid to an α-keto acid.
Amino Acid + α-Ketoglutarate ⇌ α-Keto Acid + Glutamate
[Transaminase / PLP]
Key Points:
- Enzyme: Aminotransferase (Transaminase)
- Coenzyme: PLP (Pyridoxal Phosphate = Vitamin B₆ derivative)
- All amino acids except Lysine and Threonine undergo transamination
- Reactions are reversible — used in both catabolism and synthesis
- Most reactions use α-ketoglutarate as the amino group acceptor → forming glutamate, which then undergoes oxidative deamination (by GDH) to release NH₄⁺ → enters urea cycle
- Most important transaminases clinically: ALT (liver marker) and AST (liver + heart marker)
SHORT ANSWER 2 — Phenylketonuria (PKU) and Alkaptonuria
A. Phenylketonuria (PKU)
| Feature | Detail |
|---|
| Enzyme Deficiency | Phenylalanine Hydroxylase (PAH) — converts Phe → Tyr |
| Cause | PAH deficiency OR tetrahydrobiopterin (BH₄) deficiency |
| Inheritance | Autosomal Recessive |
| Accumulated Metabolite | Phenylalanine → Phenylpyruvate (→ phenyllactate, phenylacetate) |
Flowchart:
Phenylalanine →[PAH - BLOCKED]→ Tyrosine (CANNOT form normally)
↓
Phenylpyruvate (transamination)
↓
Phenyllactate / Phenylacetate
↓
Excreted in urine → "mousy/musty" odor
Clinical Features:
- Intellectual disability (untreated) — phenylpyruvate is toxic to brain
- Fair skin, hair, eyes (reduced melanin — needs Tyr)
- Eczema
- Seizures
- Musty odor of urine and sweat (phenylacetate)
Screening: Guthrie test (heel-prick in neonates)
Treatment: Low phenylalanine diet (from birth); Sapropterin (BH₄) for responsive cases
B. Alkaptonuria
| Feature | Detail |
|---|
| Enzyme Deficiency | Homogentisate Oxidase (homogentisate 1,2-dioxygenase) |
| Pathway | Tyrosine catabolism → Homogentisate CANNOT be oxidized further |
| Inheritance | Autosomal Recessive |
Flowchart:
Tyrosine → Homogentisate →[Homogentisate Oxidase — BLOCKED]→ Maleylacetoacetate
↓
Accumulates → auto-oxidizes → dark pigment (ALKAPTON)
↓
┌───────────────────────────────┐
│ Urine turns dark (black/brown)│ → "Black urine disease"
│ on exposure to air │
└───────────────────────────────┘
Deposited in cartilage → OCHRONOSIS (bluish-black pigmentation)
→ Arthritic joint pain (later in life)
Clinical Features:
- Dark/black urine (classic sign)
- Ochronosis (pigmentation of cartilage, tendons, connective tissue)
- Arthritis (especially spine, large joints)
- Stains diapers of affected infants
Treatment: Low phenylalanine + low tyrosine diet; Nitisinone (NTBC) drug
SHORT ANSWER 3 — What is Porphyria?
Porphyria is a group of inherited (or acquired) metabolic disorders caused by enzyme deficiencies in the heme biosynthetic pathway, leading to accumulation of porphyrins or their precursors (ALA and PBG).
Normal: Glycine + Succinyl CoA → ALA → PBG → Porphyrins → Heme
↑
ENZYME BLOCK HERE causes accumulation
Two Main Types:
- Acute (Hepatic) Porphyrias → Accumulate ALA & PBG → Neuropsychiatric + abdominal pain
- Most important: Acute Intermittent Porphyria (AIP)
- Cutaneous Porphyrias → Accumulate porphyrins → Photosensitivity + skin lesions
- Most common: Porphyria Cutanea Tarda (PCT)
Classic triad of AIP:
- Abdominal pain (severe, colicky)
- Neuropsychiatric symptoms (anxiety, psychosis)
- Dark red/port-wine urine (on standing)
Key fact: Barbiturates and sulfonamides can precipitate acute attacks → contraindicated in porphyria patients
SHORT ANSWER 4 — Jaundice: Definition and Types
Definition
Jaundice (Icterus) is a yellowish discoloration of skin, mucous membranes, and sclera due to elevated serum bilirubin levels (>1.5–2 mg/dL). It is clinically detectable when total bilirubin exceeds ~3× normal (~1.5 mg/dL).
Types of Jaundice
JAUNDICE
│
┌────────────────┼────────────────┐
▼ ▼ ▼
PRE-HEPATIC HEPATIC POST-HEPATIC
(Hemolytic) (Hepatocellular) (Obstructive)
| Feature | Pre-hepatic (Hemolytic) | Hepatic (Hepatocellular) | Post-hepatic (Obstructive) |
|---|
| Cause | Excessive RBC hemolysis | Liver cell damage | Bile duct obstruction |
| Examples | Malaria, sickle cell, thalassemia | Viral hepatitis, cirrhosis | Gallstones, pancreatic cancer |
| Bilirubin type ↑ | Unconjugated ↑↑ | Both types ↑ | Conjugated ↑↑ |
| Urine bilirubin | Absent (Acholuric jaundice) | Present | Present (dark urine) |
| Urine urobilinogen | ↑↑ (increased) | ↑ | Absent (total obstruction) |
| Stool color | Dark (↑ stercobilin) | Pale/normal | Pale/clay-colored |
| Pruritus | Absent | Variable | Present (bile salts in skin) |
| ALT/AST | Normal | ↑↑↑ | Mildly ↑ |
| ALP | Normal | Mildly ↑ | ↑↑↑ |
| Van den Bergh | Indirect positive | Both | Direct positive |
Neonatal Jaundice (Physiological Jaundice):
- Appears at day 2–3; resolves by day 10
- Cause: Immature UDP-glucuronyl transferase + rapid hemolysis of fetal RBCs
- Unconjugated bilirubin ↑ → can cross BBB → Kernicterus (brain damage) if severe
SHORT ANSWER 5 — Normal Blood Urea Level & Conditions of Elevated Blood Urea
Normal Blood Urea Level:
| Parameter | Normal Value |
|---|
| Blood Urea Nitrogen (BUN) | 7–20 mg/dL |
| Serum Urea | 15–45 mg/dL (varies by lab) |
| Plasma Ammonia | 30–60 µmol/L |
Note: BUN × 2.14 = Serum Urea (approximately)
Elevated Blood Urea = UREMIA / AZOTEMIA
Two Important Conditions Where Blood Urea is Elevated:
1. Renal Failure (Kidney Disease)
Damaged kidneys → ↓ GFR → Cannot filter/excrete urea → Urea accumulates in blood
→ UREMIA (symptoms: nausea, vomiting, fatigue, confusion, "uremic frost")
- Pre-renal azotemia: Dehydration, heart failure → ↓ blood flow to kidneys
- Renal azotemia: Glomerulonephritis, pyelonephritis, diabetic nephropathy
- Post-renal azotemia: Obstruction (stones, prostate enlargement)
2. High Protein Intake / Increased Protein Catabolism
↑ Protein intake → ↑ Amino acid catabolism → ↑ NH₄⁺ → ↑ Urea synthesis → ↑ Blood urea
Also: Fever, burns, starvation, trauma → ↑ tissue protein breakdown → ↑ urea
Summary Table:
| Condition | Mechanism | BUN |
|---|
| Chronic Kidney Disease | ↓ GFR → ↓ urea excretion | ↑↑↑ |
| Acute Renal Failure | Sudden ↓ kidney function | ↑↑ |
| High-protein diet | ↑ amino acid catabolism → ↑ urea synthesis | Mildly ↑ |
| Dehydration | ↓ renal perfusion + hemoconcentration | ↑ |
| GI hemorrhage | Blood protein digested in gut → NH₄⁺ absorbed | ↑ |
| Severe burns/trauma | ↑ protein catabolism | ↑ |
| Liver failure | ↓ urea synthesis → ↑ NH₄⁺ (opposite — urea may be LOW) | ↓ or Normal |
📝 Quick Revision Summary Table
| Topic | Key Enzyme | Key Metabolite | Disease |
|---|
| Urea Cycle | CPS-I (rate-limiting) | Urea | Hyperammonemia, OTC deficiency |
| Transamination | ALT, AST (PLP needed) | Glutamate | Hepatitis, MI, alcoholic liver disease |
| Bile Pigments | UDP-Glucuronyl Transferase | Bilirubin | Jaundice, Crigler-Najjar, Gilbert, Dubin-Johnson |
| Porphyria | ALA Synthase (rate-limiting) | ALA, PBG, Porphyrins | AIP, PCT, CEP |
| PKU | Phenylalanine Hydroxylase | Phenylpyruvate | Intellectual disability, fair skin |
| Alkaptonuria | Homogentisate Oxidase | Homogentisate | Black urine, Ochronosis |
Sources: Lippincott Illustrated Reviews: Biochemistry, 8th ed.; Basic Medical Biochemistry: A Clinical Approach, 6th ed.; Harper's Illustrated Biochemistry, 32nd ed.; Guyton & Hall Textbook of Medical Physiology; Henry's Clinical Diagnosis and Management by Laboratory Methods