Phakomatoses for radiology resident
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
Reading File
Loading Image
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive, textbook-sourced content with real MRI images. Let me compile the full radiology-focused guide on phakomatoses.
Phakomatoses - Radiology Resident Guide
Phakomatoses (neurocutaneous syndromes) are congenital disorders affecting structures of ectodermal origin - the nervous system, skin, and eye. They result in hamartomas and tumours in multiple organs. The five most commonly tested in radiology are: NF-1, NF-2, Tuberous Sclerosis, von Hippel-Lindau disease, and Sturge-Weber syndrome.
1. Neurofibromatosis Type 1 (NF-1)
Genetics: Autosomal dominant. Chromosome 17q11.2 - encodes neurofibromin (tumour suppressor). Most common inherited tumour syndrome; incidence 1:3,000-4,000. 50% are new mutations.
Diagnostic Criteria (≥2 required):
| Major Criteria | Radiological? |
|---|---|
| ≥6 café-au-lait spots (>5 mm prepubertal, >15 mm postpubertal) | No |
| Axillary/inguinal freckling (Crowe sign) | No |
| ≥2 neurofibromas OR ≥1 plexiform neurofibroma | Yes |
| Optic pathway glioma | Yes |
| ≥2 Lisch nodules (iris hamartomas) | No |
| Sphenoid dysplasia / long bone cortical thinning ± pseudoarthrosis | Yes |
| First-degree relative with NF-1 | No |
CNS Imaging Findings
"Unidentified Bright Objects" (UBOs) / Neurofibromatosis Bright Objects (NBOs):
- Present in 60-80% of NF-1 children; up to 95% with coexistent OPG
- T2/FLAIR hyperintense foci, no mass effect, no enhancement
- Typical sites: pons, cerebellar white matter, internal capsules, basal ganglia (may be slightly T1 hyperintense here), thalami, hippocampi
- Age-dependent: rare <4 years, peak 4-10 years, virtually absent after age 20
- Key distinction from glioma: no growth, no enhancement; enhancement or increasing mass effect should raise concern for tumour transformation
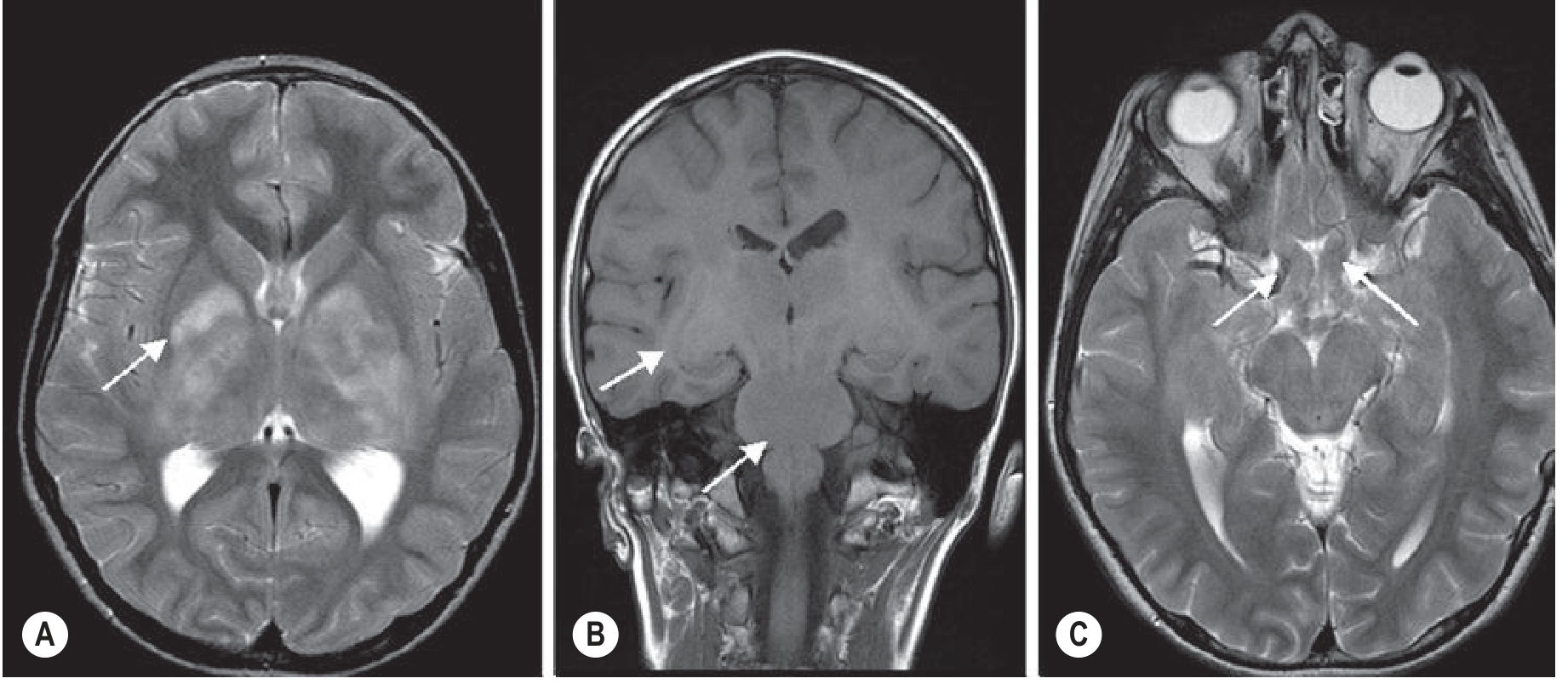
Optic Pathway Gliomas (OPGs):
- Most common brain abnormality in NF-1 (up to 15%); usually WHO grade I pilocytic astrocytoma
- NF-1 OPGs more often affect optic nerves (vs. chiasm/postchiasmatic in sporadic cases); better prognosis
- Fusiform optic nerve expansion; widening of optic foramen on CT
- MRI preferred: fat-suppressed T1+Gd and STIR sequences; 3 mm slices
- Once chiasm/hypothalamus involved: risk of precocious puberty and visual loss
Plexiform Neurofibromas:
- Major diagnostic criterion; multinodular, involve multiple nerve fascicles
- Classic site: orbit along ophthalmic V1, associated with sphenoid wing dysplasia
- CT: hypodense, minimal enhancement
- MRI: T1 low SI, T2 heterogeneous hyperintense; variable Gd enhancement; "target sign" (central T2 hypointensity) in neurofibromas
- Extension into pterygomaxillary fissure, orbital apex, cavernous sinus
- Malignant transformation to fibrosarcoma: 2-12%
Sphenoid Wing Dysplasia:
- Produces the "bare/empty orbit" sign on plain radiograph
- Temporal lobe herniation through the orbit
- Pulsatile exophthalmos (CSF pulsations transmitted through defect)
Other NF-1 CNS tumours: Brainstem gliomas (medulla/midbrain > pons - unusual for NF-1), cerebellar pilocytic astrocytomas (1-3%), tectal gliomas (can cause aqueductal stenosis + hydrocephalus)
Skeletal: Kyphoscoliosis (high thoracic acute curve), tibial bowing/pseudoarthrosis, lambdoid sutural dysplasia, rib notching (dumbbell neurofibromas), focal gigantism
2. Neurofibromatosis Type 2 (NF-2)
Genetics: Autosomal dominant. Chromosome 22q12 - encodes merlin/schwannomin. Incidence 1:50,000.
Diagnostic Criteria:
- Bilateral vestibular schwannomas (pathognomonic) - shown on MRI/CT or histology
- OR first-degree relative with NF-2 + unilateral 8th nerve tumour
- OR first-degree relative with NF-2 + any two of: neurofibroma, meningioma, schwannoma, glioma, or juvenile posterior subcapsular lens opacity
Key Imaging Features:
- Bilateral vestibular (acoustic) schwannomas - enhancing masses at the internal auditory canals (IAC); adults present with hearing loss; children more often with seizures or facial palsy
- Multiple meningiomas (often en plaque)
- Cranial nerve schwannomas (CN V, VII common)
- Ependymomas (especially spinal; cervical cord)
- Spinal schwannomas on cauda equina roots - dumbbell morphology, foraminal widening
- MRI protocol: Contrast-enhanced T1 with thin cuts through posterior fossa; FIESTA/CISS sequences for IAC; full spine survey
3. Tuberous Sclerosis Complex (TSC)
Genetics: Autosomal dominant (66-86% spontaneous mutations). Two genes:
- TSC1 - chromosome 9q34.3 - hamartin
- TSC2 - chromosome 16p13.3 - tuberin (adjacent to PKD1; more severe phenotype, more SEN nodules, renal angiomyolipomas)
- Both products form a functional unit regulating the mTOR pathway
Prevalence: 1:6,000
CNS Imaging Findings
Cortical Tubers:
- Hamartomatous cortical dysplasias; hallmark lesion; cause epilepsy
- CT: often calcified (higher density); may be occult early
- MRI: T2/FLAIR hyperintense; involves cortex + subcortical white matter; signal may invert (become T1 bright) as child matures
Subependymal Nodules (SENs):
- Project into ventricles along the caudate eminences ("candle drippings" appearance)
- Calcify with age → T2 hypointense on MRI (hyperdense on CT)
- Stable, non-enhancing; typically benign
Subependymal Giant Cell Astrocytomas (SEGAs):
- Most important complication: arise from SENs at/near the foramen of Monro
- Enhancing mass; causes obstructive hydrocephalus
- WHO grade I tumour; surgical or mTOR inhibitor (everolimus) treatment
- MRI: T1/T2 heterogeneous, avid Gd enhancement; size >1 cm and growth are key criteria
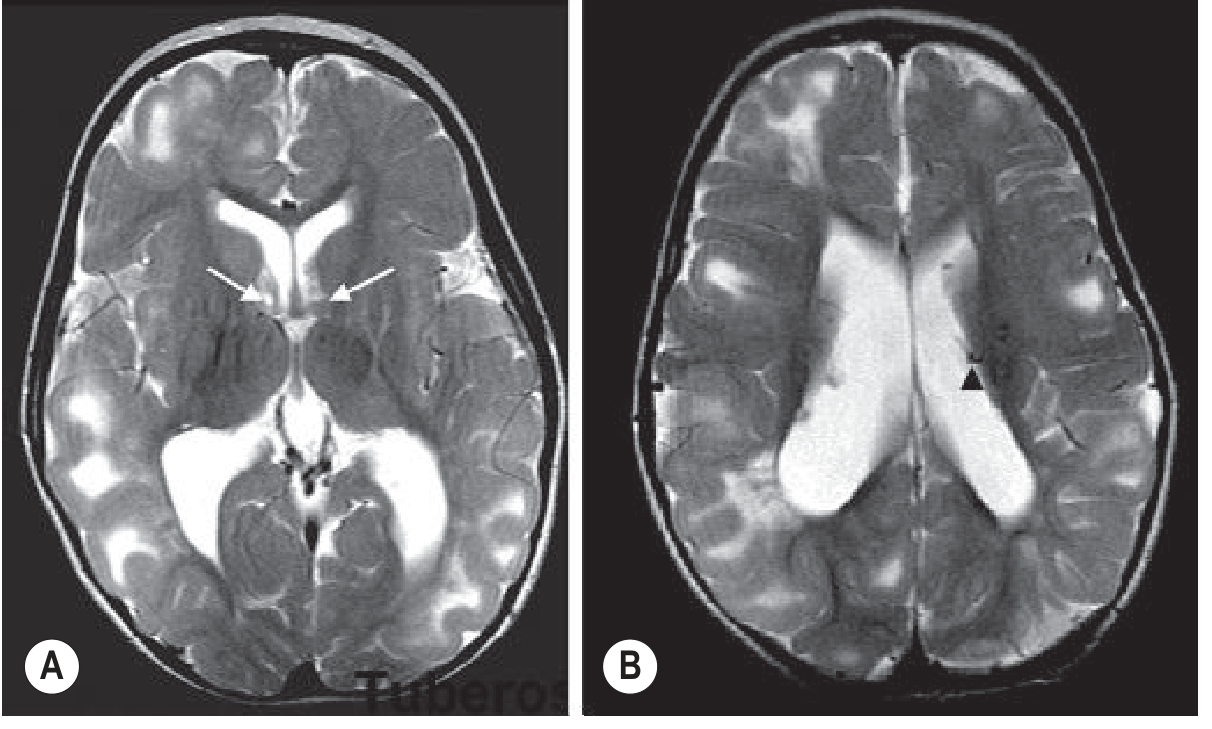
Radial Migration Lines (White Matter Lesions):
- T2 hyperintense lines extending from ventricle to cortex
- Reflect abnormal neuronal migration; not visible on CT
Systemic Imaging Findings
| Organ | Finding |
|---|---|
| Kidney | Angiomyolipomas (bilateral, multiple; fat-containing - T1 bright, loss of signal on fat-sat; CT: fat attenuation), renal cysts, renal cell carcinoma |
| Lung | Pulmonary LAM (lymphangioleiomyomatosis) - thin-walled cysts, predominantly in women; chylous effusion |
| Heart | Cardiac rhabdomyomas - echogenic masses; most regress spontaneously; can cause arrhythmia |
| Skeleton | Sclerotic bone islands (vertebral bodies, pedicles); irregular periosteal reaction (metacarpals); cyst-like phalangeal lesions |
| Eye | Retinal astrocytic hamartomas ("mulberry lesion") |
4. Von Hippel-Lindau (VHL) Disease
Genetics: Autosomal dominant. Chromosome 3p25 - VHL tumour suppressor gene (regulates HIF/VEGF pathway). Prevalence ~1:40,000.
Classification:
- Type 1 (no phaeochromocytoma): haemangioblastomas + renal/pancreatic lesions
- Type 2 (with phaeochromocytoma): subdivided by presence of RCC
Imaging Findings
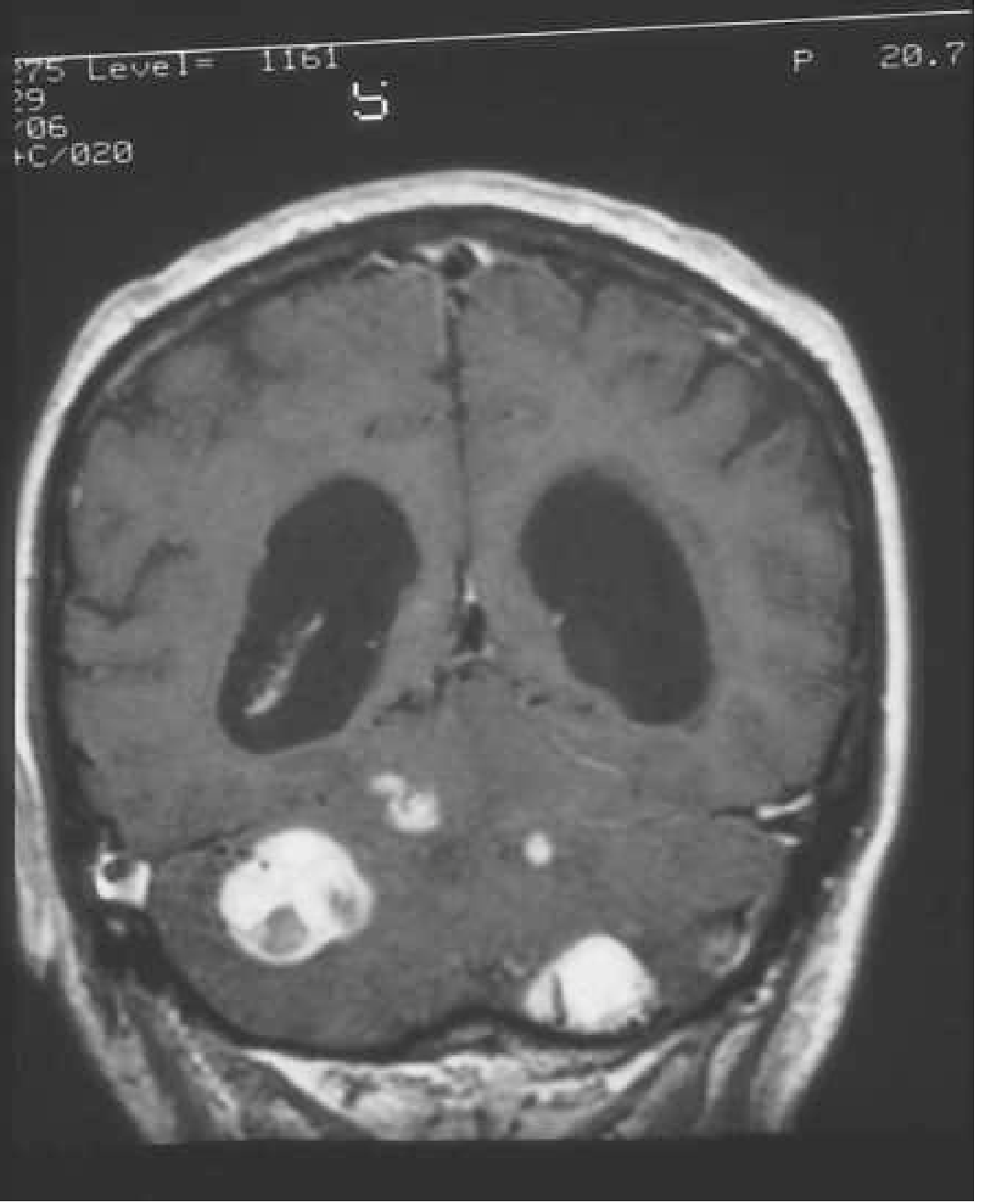
CNS Haemangioblastomas (most important):
- Benign, slow-growing vascular tumours
- Sites: cerebellum (~50%, hemispheres >> vermis), spinal cord (intramedullary), medulla (area postrema)
- Classic MRI appearance: cyst with enhancing mural nodule (nodule is avid, cyst wall does not enhance)
- Solid variants occur (more common in brainstem)
- Spinal haemangioblastomas → syringomyelia (common)
- Screening: contrast-enhanced MRI brain + spine (pre + post T1 with thin posterior fossa cuts)
Retinal Angiomas: Reddish masses with feeding artery and draining vein; treated with photocoagulation
Renal Cell Carcinoma: Clear cell type; bilateral, multifocal; hypervascular on CT/MRI
Phaeochromocytoma: Bilateral in ~40%; T2 hyperintense "lightbulb" on MRI; avid enhancement; catecholamine excess
Pancreatic lesions: Simple cysts (most common), serous cystadenomas, pancreatic NETs (hypervascular)
Endolymphatic sac tumours (ELSTs): 10-15% of VHL; petrous bone; papillary appearance; T1 hyperintense areas (haemorrhage); cause hearing loss and tinnitus
5. Sturge-Weber Syndrome
Genetics: Sporadic; somatic activating mutation in GNAQ (disrupts G-protein alpha subunit GTPase activity). NOT inherited.
Classic Triad:
- Port-wine naevus (facial capillary haemangioma - forehead/upper eyelid distribution; V1 territory)
- Ipsilateral leptomeningeal angioma (parieto-occipital predominance)
- Glaucoma (ipsilateral; two age peaks: infancy and late childhood)
Only 10-20% of children with a port-wine naevus develop an intracranial angioma.
Imaging Findings
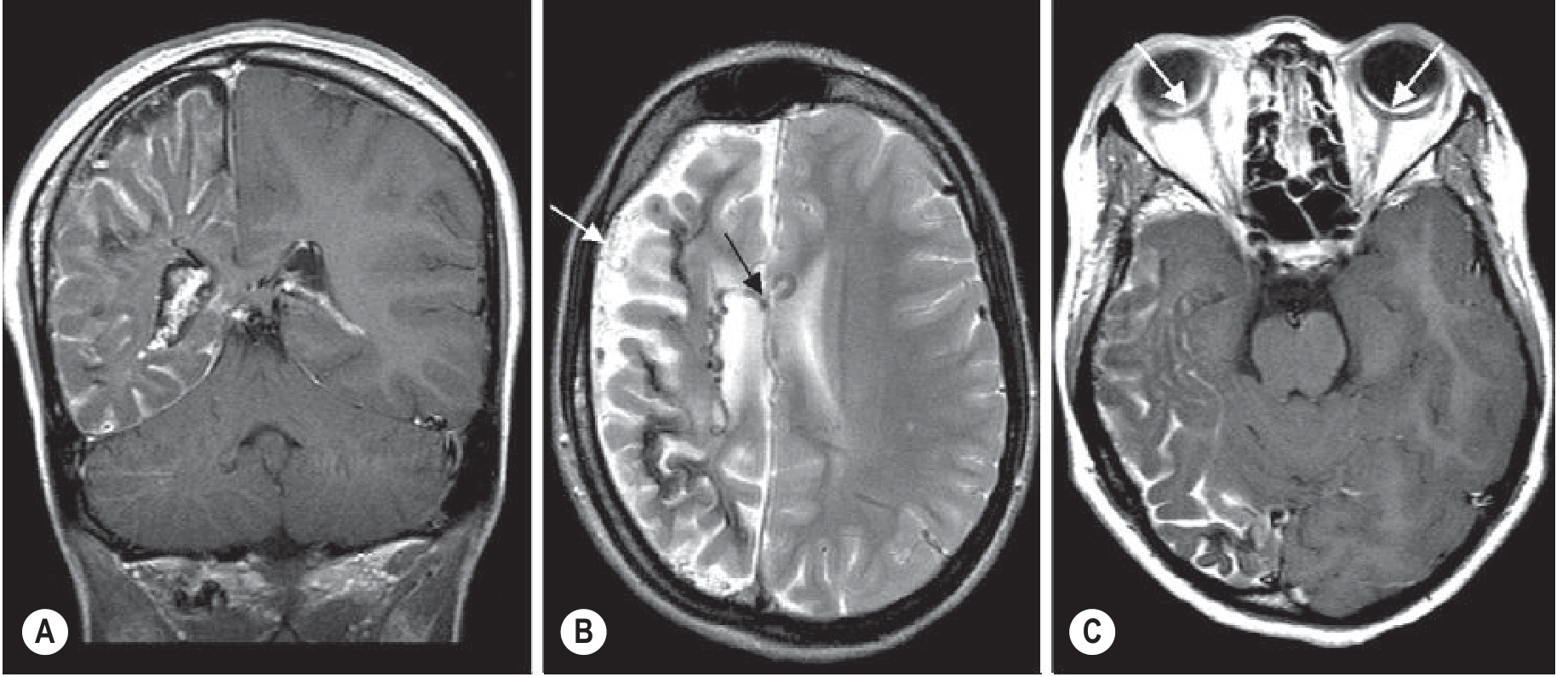
CT:
- "Tramline calcifications" - gyriform cortical calcifications (visible by ~2 years)
- Leptomeningeal angioma may not be visible without contrast in early life
- Ipsilateral hemispheric atrophy in established disease
MRI:
- Diffuse pial enhancement over affected hemisphere (variable thickness) - most sensitive finding
- Underlying cortical atrophy; T2 hypointensity within gyri (calcification)
- Ipsilateral choroid plexus enlargement - characteristic, due to collateral venous drainage
- Dilated transparenchymal medullary veins (T2 hypointense flow voids) communicating superficial-deep venous systems
- Bilateral involvement in ≥15% of patients (including some with unilateral nevus)
- "Burnt-out" cases: pial angioma may no longer enhance; shrunken calcified hemisphere remains
- Polymicrogyria may coexist
Quick-Reference Comparison Table
| Feature | NF-1 | NF-2 | TSC | VHL | Sturge-Weber |
|---|---|---|---|---|---|
| Gene/Chr | NF1 / 17q | NF2 / 22q | TSC1(9q), TSC2(16p) | VHL / 3p | GNAQ (somatic) |
| Inheritance | AD | AD | AD | AD | Sporadic |
| Pathognomonic CNS lesion | UBOs + OPG | Bilateral vestibular schwannomas | Cortical tubers + SEGAs | Cerebellar haemangioblastoma | Pial angioma |
| Key CT finding | Sphenoid dysplasia, "bare orbit" | CPA mass bilateral | Calcified SEN, tubers | - | Tramline calcifications |
| Key MRI finding | T2 UBOs (basal ganglia, pons); fusiform optic nerve | Bilateral IAC enhancing masses | Tubers, SEN, SEGAs at foramen of Monro | Cyst + enhancing mural nodule (cerebellum) | Pial enhancement, enlarged choroid plexus |
| Malignant risk | Fibrosarcoma (NF → 2-12%); MPNST | Low | RCC (TSC2), SEGA (benign) | RCC (clear cell), phaeo | Low |
| Skin | Café-au-lait, neurofibromas | Few | Ash-leaf spots, adenoma sebaceum, shagreen patch | - | Port-wine naevus (V1) |
| Eye | Lisch nodules | Posterior subcapsular cataract | Retinal astrocytic hamartoma | Retinal angioma | Glaucoma, choroidal angioma |
| Kidney | - | - | AML (fat-containing), cysts | RCC (bilateral, multifocal), cysts | - |
| Spine | Dumbbell neurofibromas, scoliosis | Ependymomas, schwannomas | - | Haemangioblastomas + syrinx | - |
Additional / Less Common Phakomatoses
Neurocutaneous Melanosis:
- Giant congenital melanocytic naevi (skin) + intracranial melanosis
- MRI: T1 shortening (melanin) in anterior/mesial temporal lobe, cerebellum, pons
- CT: areas of increased density (less conspicuous)
- Hydrocephalus from leptomeningeal involvement; rare malignant transformation
Wyburn-Mason Syndrome:
- Racemose (arteriovenous) malformations in retina
- Ipsilateral intracranial AVM (mid-brain/optic pathway); no treatment required unless symptomatic
PHACES Syndrome:
- Posterior fossa malformations (Dandy-Walker), facial Haemangiomas, Arterial anomalies, Cardiac/eye anomalies, Sternal cleft
Basal Cell Naevus Syndrome (Gorlin Syndrome):
- Multiple basal cell carcinomas, odontogenic keratocysts
- Calcification of falx cerebri (pathognomonic on CT/skull XR)
- Medulloblastoma (desmoplastic type); bifrontal cortical dysplasia
Key Radiology Pearls
- NF-1 UBOs: T2 bright, no enhancement, no mass effect → watch for change; if they enhance = suspect glioma
- NF-1 "bare orbit": Sphenoid wing dysplasia on plain film/CT
- TSC SEGAs: Grow at the foramen of Monro → obstructive hydrocephalus; any SEN >1 cm + enhancing = SEGA
- TSC AML: Fat attenuation on CT (<-10 HU); spontaneous retroperitoneal haemorrhage ("Wunderlich syndrome") if >4 cm
- VHL haemangioblastoma: Cyst + mural nodule in cerebellum of young adult → first think VHL (especially if multiple or spinal)
- Sturge-Weber: Early MRI may be normal - pial enhancement is the earliest MRI sign, calcifications come later; enlarged ipsilateral choroid plexus is characteristic
- NF-2: Any bilateral IAC masses = NF-2 until proven otherwise
Sources: Grainger & Allison's Diagnostic Radiology (Table 76.2, Figs. 76.27-76.32); Bradley & Daroff's Neurology in Clinical Practice (Boxes 99.1-99.2, Chapter 99)
This is a shared conversation. Sign in to Orris to start your own chat.