Gauchers disease in detail
Reading File
Finding Sources
Searching PubMed
"Gaucher disease"[MeSH Terms] AND treatment
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have extremely comprehensive content to write a detailed, well-organized answer. Let me compile it all.
Gaucher Disease - A Detailed Overview
Gaucher disease is the most common lysosomal storage disorder and the most frequent inherited sphingolipidosis. It is caused by the autosomal recessive deficiency of the enzyme glucocerebrosidase (beta-glucosidase), resulting in the pathological accumulation of glucocerebroside (glucosylceramide) within macrophages throughout the body.
1. Genetics and Molecular Basis
-
Gene: GBA1 (formerly GBA), located on chromosome 1q21
-
Inheritance: Autosomal recessive
-
Enzyme deficient: Lysosomal glucocerebrosidase (acid beta-glucosidase)
-
Substrate accumulated: Glucocerebroside (glucosylceramide), derived from degradation of senescent erythrocytes and leukocytes
-
Over 600 different mutations identified; key ones include:
- N370S (p.N409S): Most common allele in Ashkenazi Jews; found exclusively in Type 1 - protects against neurologic involvement
- L444P (p.L483P): Homozygosity most often associated with Type 3
- D409H (p.D448H): Homozygosity defines Type 3C (with cardiac calcifications and hydrocephalus)
-
A highly homologous pseudogene nearby complicates molecular diagnosis - specialized lab knowledge is required
-
The GBA1 locus is also the most common genetic risk factor for Parkinson disease identified to date; both affected patients and heterozygous carriers have a 5- to 10-fold increased frequency of parkinsonism
-
Goldman-Cecil Medicine, p. 2303; Robbins & Kumar Basic Pathology, p. 3656
2. Epidemiology
-
Pan-ethnic disorder, but type 1 shows marked enrichment in Ashkenazi Jews:
- Carrier frequency in Ashkenazi Jews: ~1 in 16
- General population carrier frequency: ~1 in 100 (Robbins notes up to 1 in 40,000 for non-Jewish populations for the type 1 variant)
-
Type 1 accounts for approximately 99% of all cases globally
-
Types 2 and 3 are rare and more severe
-
Goldman-Cecil Medicine, p. 2303; Robbins & Kumar Basic Pathology, p. 3658
3. Pathophysiology
The enzyme glucocerebrosidase normally degrades glucocerebroside (a glycolipid intermediate from breakdown of red and white blood cell membranes). Its deficiency leads to:
- Accumulation of glucocerebroside within the lysosomes of macrophages (particularly in liver, spleen, and bone marrow)
- Gaucher cells - the pathognomonic enlarged macrophages (up to 100 µm diameter) with lipid-distended lysosomes producing the classic "wrinkled tissue paper" or "crumpled silk" cytoplasm
- Macrophage activation: elevated cytokines (IL-1, IL-6, TNF) drive bone disease and organomegaly - not simply a passive storage phenomenon
- Glucosylsphingosine (lyso-Gb1) also accumulates and is neurotoxic; it is the basis for the serum/blood biomarker assay increasingly used in diagnosis
Histology of Gaucher cells:
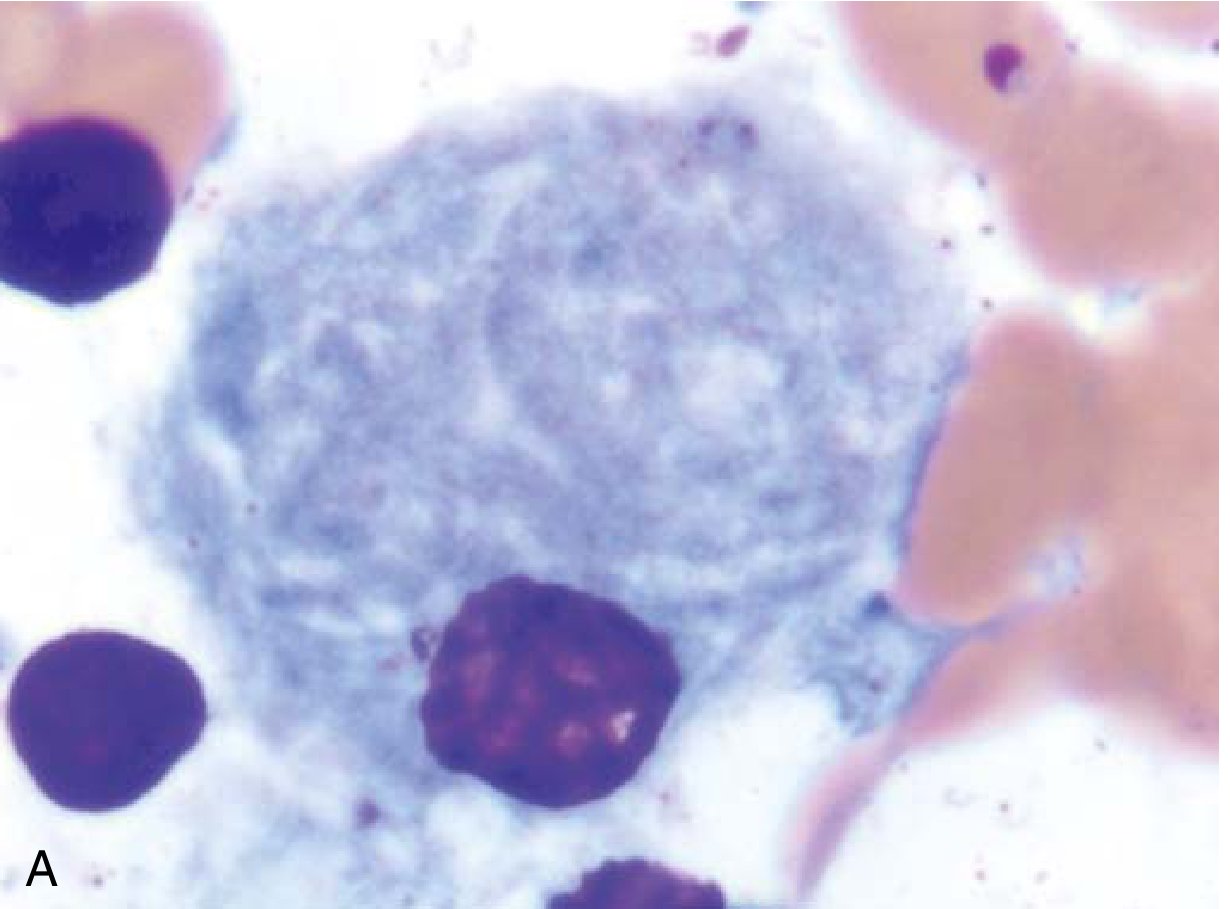
Gaucher cell in bone marrow aspirate: an enlarged macrophage with characteristic "wrinkled tissue paper" cytoplasm - Robbins & Kumar Basic Pathology
- Robbins & Kumar Basic Pathology, p. 3656; Thompson & Thompson Genetics, p. 787
4. Classification: Three Types
| Feature | Type 1 | Type 2 | Type 3 |
|---|---|---|---|
| Name | Chronic non-neuronopathic | Acute neuronopathic | Subacute/chronic neuronopathic |
| Frequency | ~99% of cases | Rare | Rare |
| CNS involvement | Absent | Severe, rapid | Present but variable |
| Onset | Any age (often adult) | Perinatal/infancy | Childhood |
| Prognosis | Compatible with long life | Death by age 2 | Variable; reduced lifespan |
| Common mutation | N370S | Various | L444P homozygous; D409H (3C) |
5. Clinical Manifestations
Type 1 (Non-neuronopathic)
- Hepatosplenomegaly: Painless splenomegaly is the most common presentation; spleen may be massively enlarged, filling the entire abdomen
- Hematologic: Thrombocytopenia, anemia (from hypersplenism and marrow replacement), easy bruising, epistaxis
- Bone disease (most significant cause of morbidity):
- Classic Erlenmeyer flask deformity of the distal femur (cortical thinning, widening of the medullary cavity)
- Osteopenia, lytic bone lesions, osteoporosis, pathologic fractures (hip, ribs, spine)
- Bone crises: Episodic severe bone pain from infarction, lasting weeks
- Aseptic necrosis of femoral heads
- Laboratory findings: Anemia, thrombocytopenia, elevated ferritin, elevated acid phosphatase, elevated ACE, elevated lyso-Gb1, elevated liver enzymes
- Immunologic: ~50% develop polyclonal gammopathy; ~30% develop monoclonal gammopathy; increased risk of multiple myeloma
- Pulmonary: Occasional pulmonary hypertension
- Parkinsonism risk: Significantly elevated (5- to 10-fold) compared to the general population
Type 2 (Acute neuronopathic)
- Onset at or shortly after birth
- Rapid neurodegeneration: failure to thrive, laryngospasm, strabismus, seizures
- Extensive visceral involvement
- Death within the first 2 years of life
Type 3 (Subacute neuronopathic)
- Onset in childhood, variable course
- Organomegaly and bone involvement (similar to type 1)
- Abnormal horizontal saccadic eye movements - hallmark neurologic sign
- Some develop myoclonic epilepsy or neurodegeneration
- Type 3C (D409H homozygous): cardiac valve calcifications, hydrocephalus, corneal opacities
Radiographic findings:
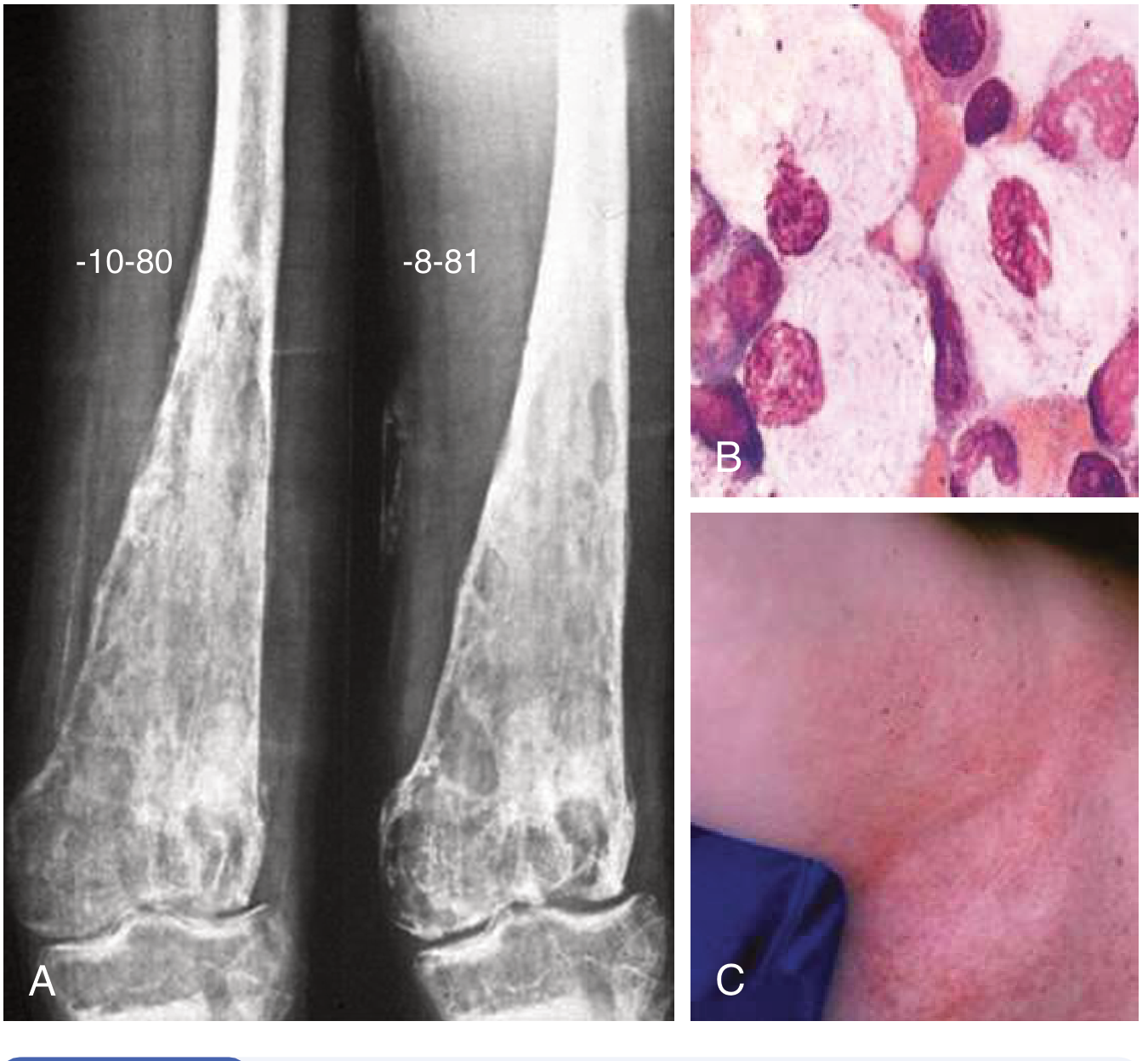
A: Erlenmeyer flask deformity - cortical thinning and widening of the metaphysis and adjacent diaphysis. B: Gaucher cells showing lipid storage. C: Angiokeratomas in Fabry disease (for comparison). - Goldman-Cecil Medicine
- Goldman-Cecil Medicine, pp. 2302-2304; Robbins & Kumar Basic Pathology, pp. 3656-3658
6. Diagnosis
Approach (Table 192-4, Goldman-Cecil):
Suspect Gaucher disease when a patient has:
- Unexplained organomegaly (hepatosplenomegaly)
- Thrombocytopenia/anemia with no clear cause
- Painful bone crises or pathologic fractures
- Erlenmeyer flask deformity on X-ray
- Abnormal saccadic eye movements
- Family history of Gaucher disease
- Unexplained multiple myeloma or parkinsonism
Diagnostic steps:
- Biomarker screen: Lyso-Gb1 (glucosylsphingosine) - measured in dried blood spot or serum; massively elevated and increasingly used as first-line screen
- Enzyme assay: Deficient glucocerebrosidase activity in leukocytes or cultured fibroblasts - confirms diagnosis
- Molecular analysis: Full GBA1 gene sequencing - needed for accurate molecular diagnosis (screening for N370S covers ~70% of Ashkenazi Jewish alleles)
- Bone marrow/liver biopsy: Shows Gaucher cells but not indicated for diagnosis
- Carrier identification: DNA testing when mutant allele is known
- Prenatal diagnosis: Enzyme activity or mutation testing on chorionic villi or amniotic fluid
- Goldman-Cecil Medicine, pp. 2303-2304
7. Treatment
Enzyme Replacement Therapy (ERT)
The primary treatment for Type 1 and Type 3 Gaucher disease:
| Agent | Details |
|---|---|
| Imiglucerase (Cerezyme) | Plant cell-derived recombinant glucocerebrosidase |
| Velaglucerase alfa (VPRIV) | Human cell line-derived |
| Taliglucerase alfa (Elelyso) | Carrot cell-derived |
- Dose: 15-60 IU/kg IV every 2 weeks
- Response: Anemia, thrombocytopenia, and organomegaly reverse within 12-36 months
- Limitation: Does NOT cross the blood-brain barrier - does not halt neurologic progression in Types 2/3
- Enzyme is targeted to macrophage lysosomes via mannose-6-phosphate or mannose residues
- Treatment is ongoing, lifelong, and very expensive
Substrate Reduction Therapy (SRT)
Reduces substrate synthesis to match the impaired degradation rate:
- Eliglustat (Cerdelga): Ceramide analogue; potent specific inhibitor of glucosylceramide synthase
- Administered orally
- Significantly improves spleen/liver volumes, hemoglobin, and platelet counts
- Preferred by some adults over IV ERT
- Important: Check CYP2D6 metabolizer status before use
- Ultra-rapid metabolizers: NOT eligible
- Normal or intermediate metabolizers: 84 mg twice daily
- Poor metabolizers: 84 mg once daily
- Miglustat (Zavesca): Earlier oral SRT agent; less preferred due to side effect profile
Supportive Therapies
- Bisphosphonates for osteoporosis
- Orthopedic surgery for fractures; joint replacement for avascular necrosis
- Aggressive pain management and hydration for bone crises
- Splenectomy: Now rarely indicated (used before ERT was available; accelerates bone disease)
- Hematopoietic stem cell transplantation: Can improve systemic but not neurologic manifestations; reserved for severe disease in settings where ERT/SRT are unavailable
Emerging/Investigational Therapies
-
Brain-penetrant ERT and SRT (for neuronopathic types)
-
Chemical chaperones: e.g., ambroxol (high-dose) - a recent 2024 systematic review [PMID 39116528] investigated this approach for Types 2 and 3, showing preclinical and clinical promise; it stabilizes misfolded glucocerebrosidase
-
Gene therapy: Active area of research
-
Standard therapies do not prevent the development of parkinsonism
-
Goldman-Cecil Medicine, pp. 2303-2304; Thompson & Thompson Genetics, p. 792-794; Emery's Medical Genetics, p. 1732
8. Gaucher Disease and Parkinson Disease
This is an increasingly important association:
- Both patients with Gaucher disease and heterozygous GBA1 carriers have elevated parkinsonism risk
- GBA1 mutations are found in 5-10% of all Parkinson disease patients worldwide - making it the single most common genetic risk factor for parkinsonism
- Mechanism likely involves impaired lysosomal function, alpha-synuclein accumulation, and Lewy body formation
- Reduced penetrance means the majority of Gaucher patients and carriers do not develop Parkinson disease
- A 2025 MDSGene systematic review [PMID 39927608] further classified GBA1 variants and their genotype-phenotype relationships in parkinsonism
9. Summary Table
| Parameter | Detail |
|---|---|
| Category | Lysosomal storage disease (sphingolipidosis) |
| Enzyme deficient | Glucocerebrosidase (acid beta-glucosidase) |
| Gene | GBA1, chromosome 1q21 |
| Substrate stored | Glucocerebroside in macrophages |
| Pathognomonic cell | Gaucher cell ("wrinkled tissue paper" cytoplasm) |
| Most common type | Type 1 (non-neuronopathic, ~99%) |
| Most common presentation | Splenomegaly, thrombocytopenia, bone disease |
| Classic X-ray sign | Erlenmeyer flask deformity |
| Diagnosis | Leukocyte glucocerebrosidase activity + GBA1 sequencing |
| Biomarker | Lyso-Gb1 (glucosylsphingosine) |
| First-line treatment | ERT (imiglucerase/velaglucerase) or oral eliglustat |
| Treatment limitation | ERT does not cross the BBB |
| Parkinson risk | 5-10x elevated; GBA1 = most common genetic PD risk factor |
Recent evidence note: A 2025 systematic review (MDSGene, PMID 39927608) provided updated GBA1 variant classification and genotype-phenotype data relevant to Parkinson risk. A 2024 systematic review (PMID 39116528) assessed high-dose ambroxol as a pharmacological chaperone for neuronopathic Gaucher disease (Types 2 and 3), showing promising preclinical and limited clinical data - this is not yet standard of care.
Sources: Goldman-Cecil Medicine (International Edition), pp. 2302-2304; Robbins & Kumar Basic Pathology, pp. 3655-3658; Emery's Elements of Medical Genetics and Genomics, pp. 1724-1734; Thompson & Thompson Genetics and Genomics in Medicine, pp. 787-794
This is a shared conversation. Sign in to Orris to start your own chat.