Pathology of diabetes
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the content needed for a comprehensive answer. Here is a thorough overview of the pathology of diabetes mellitus, drawn from Robbins, Cotran & Kumar Pathologic Basis of Disease and Robbins & Kumar Basic Pathology.
Pathology of Diabetes Mellitus
Diabetes mellitus (DM) is a group of metabolic disorders sharing chronic hyperglycemia caused by defects in insulin secretion, insulin action, or both. Chronic hyperglycemia causes secondary damage to the kidneys, eyes, nerves, and blood vessels — making DM the leading cause of end-stage renal disease, adult-onset blindness, and non-traumatic lower-extremity amputations in the United States.
Diagnosis
Blood glucose is normally maintained between 70–120 mg/dL. Diagnostic criteria (ADA/WHO):
| Criterion | Threshold |
|---|---|
| Fasting plasma glucose | ≥ 126 mg/dL |
| Random plasma glucose (with symptoms) | ≥ 200 mg/dL |
| 2-hr glucose during OGTT (75 g load) | ≥ 200 mg/dL |
| HbA1c | ≥ 6.5% |
Prediabetes: FPG 100–125 mg/dL, 2-hr OGTT 140–199 mg/dL, or HbA1c 5.7–6.4%. Up to 25% develop overt T2D within 5 years.
Classification
Type 1 Diabetes (T1D) — ~5–10% of cases
- Autoimmune destruction of pancreatic β cells → absolute insulin deficiency
- Most common in patients < 20 years; nonobese
- Progressive fall in insulin levels
- Circulating islet autoantibodies (anti-GAD65, anti-IA2, anti-ZnT8)
- Risk of diabetic ketoacidosis (DKA)
Type 2 Diabetes (T2D) — ~90–95% of cases
- Combination of peripheral insulin resistance + inadequate β-cell compensatory response
- ~80% are obese; increasingly seen in adolescents
- Early hyperinsulinemia, then relative insulin deficiency
- No islet autoantibodies
- Risk of nonketotic hyperosmolar coma
Other Types
- Monogenic (MODY): Loss-of-function mutations in glucokinase (GCK/MODY2), HNF1α (MODY3), HNF4α (MODY1), PDX1, HNF1β, NEUROD1, and others
- Gestational diabetes mellitus (GDM)
- Secondary causes: pancreatitis, haemochromatosis, drug-induced, endocrinopathies (Cushing syndrome, acromegaly)
Comparison Table: T1D vs T2D
| Feature | T1D | T2D |
|---|---|---|
| Onset | Childhood/adolescence | Adult (increasingly younger) |
| Body habitus | Normal/underweight | Obese (80%) |
| Insulin levels | Progressive decrease | High early; falls late |
| Autoantibodies | Present | Absent |
| HLA linkage | Yes (HLA class II; CTLA4, PTPN22) | No |
| Islet pathology | Insulitis, β-cell depletion | Amyloid deposition, mild β-cell loss |
| Acute complication | DKA | Hyperosmolar hyperglycemic state |
Pathogenesis of T1D
T1D results from a breakdown in self-tolerance to islet antigens, mediated principally by T cells:
- Genetic susceptibility: Strong linkage to MHC class II alleles (HLA-DR3, DR4, DQ variants); polymorphisms in CTLA4 and PTPN22 impair T-cell regulation
- Environmental triggers: Viral infections (enteroviruses), gut microbiome alterations, and dietary factors may trigger islet autoimmunity in genetically susceptible individuals
- Insulitis: CD8+ and CD4+ T cells and macrophages infiltrate the islets, directly killing β cells via perforin/granzyme and Fas-FasL pathways
- Autoantibodies: Anti-islet antibodies (anti-GAD65, anti-IA2, anti-ZnT8) appear years before clinical disease and serve as biomarkers
Histology: Reduction in islet size and number; mononuclear inflammatory infiltrate (insulitis); β-cell depletion with relative preservation of α, δ, and PP cells.
Pathogenesis of T2D
Two interrelated defects:
1. Insulin Resistance
Insulin target tissues (especially skeletal muscle, liver, adipose) fail to respond normally to insulin. Key contributors:
- Obesity (especially visceral adiposity): excess free fatty acids (FFAs) activate serine kinases that phosphorylate IRS-1, impairing downstream PI3K–AKT signalling
- Adipokines: Leptin resistance, reduced adiponectin, elevated TNF-α and IL-6 from adipose tissue promote hepatic gluconeogenesis and impair glucose uptake
- Lipotoxicity: Ectopic fat deposition in muscle and liver
- Metabolic syndrome: Insulin resistance + hypertension + dyslipidaemia + central obesity
2. β-Cell Dysfunction
- Initially, β cells hyperfunction to compensate for insulin resistance
- Eventually, β cells fail due to: lipotoxicity, glucotoxicity (chronic hyperglycemia), impaired incretin effect (reduced GLP-1 and GIP secretion), and islet amyloid polypeptide (IAPP/amylin) deposition
- Islet amyloid deposition is present in >90% of long-standing T2D islets, but whether it causes or results from β-cell burnout is debated
Histology: Mild reduction in β-cell mass; amyloid deposits replacing islet cells (congophilic material — amylin aggregates); no insulitis.
Pathogenesis of Chronic Complications (Glucotoxicity)
Persistent hyperglycemia damages tissues via at least four mechanisms:
1. Advanced Glycation End Products (AGEs)
- Nonenzymatic glycation of proteins generates AGE precursors (glyoxal, methylglyoxal, 3-deoxyglucosone)
- AGEs bind RAGE receptor on endothelium, macrophages, vascular smooth muscle → release of TGF-β, VEGF, ROS, and procoagulant factors
- Cross-link extracellular matrix proteins → trap LDL in vessel walls (accelerating atherosclerosis) and albumin in capillary walls (basement membrane thickening)
2. Protein Kinase C (PKC) Activation
- Intracellular hyperglycemia increases diacylglycerol (DAG) from glycolytic intermediates → activates PKC
- Downstream: increased VEGF (retinopathy), TGF-β (fibrosis), PAI-1 (thrombosis), reduced eNOS (impaired vasodilation)
3. Hexosamine Pathway
- Excess glucose shunted into hexosamine pathway → O-GlcNAc modification of transcription factors → abnormal gene expression, impaired insulin signalling
4. Polyol Pathway
- Aldose reductase converts glucose to sorbitol (consumes NADPH) → oxidative stress, depletion of glutathione → endothelial damage (especially in lens and peripheral nerves)
Morphologic Changes (Organ Pathology)
Pancreas
- T1D: Reduced islet number and size; insulitis (lymphocytic infiltrate); β-cell depletion; α, δ, and PP cells preserved
- T2D: Mild β-cell reduction; islet amyloid (amylin deposits, Congo red positive)
Vascular System
- Macrovascular disease (large and medium arteries): Accelerated atherosclerosis → myocardial infarction, stroke, peripheral arterial disease
- Microvascular disease (capillaries and arterioles): Basement membrane thickening; hyaline arteriolosclerosis (especially afferent and efferent renal arterioles); pericyte loss
Kidneys — Diabetic Nephropathy
Three cardinal lesions:
- Glomerular lesions: Diffuse glomerulosclerosis (most common — diffuse increase in mesangial matrix); nodular glomerulosclerosis (Kimmelstiel-Wilson nodules) — ovoid PAS-positive deposits in the mesangium, pathognomonic of diabetic nephropathy
- Vascular lesions: Hyaline arteriolosclerosis of both afferent and efferent arterioles (efferent arteriole involvement is virtually diagnostic of diabetes)
- Tubular lesions: Glycogen accumulation in tubular epithelial cells ("Armanni-Ebstein lesion"); thickening of tubular basement membranes
- Progression: microalbuminuria → macroalbuminuria → declining GFR → ESRD
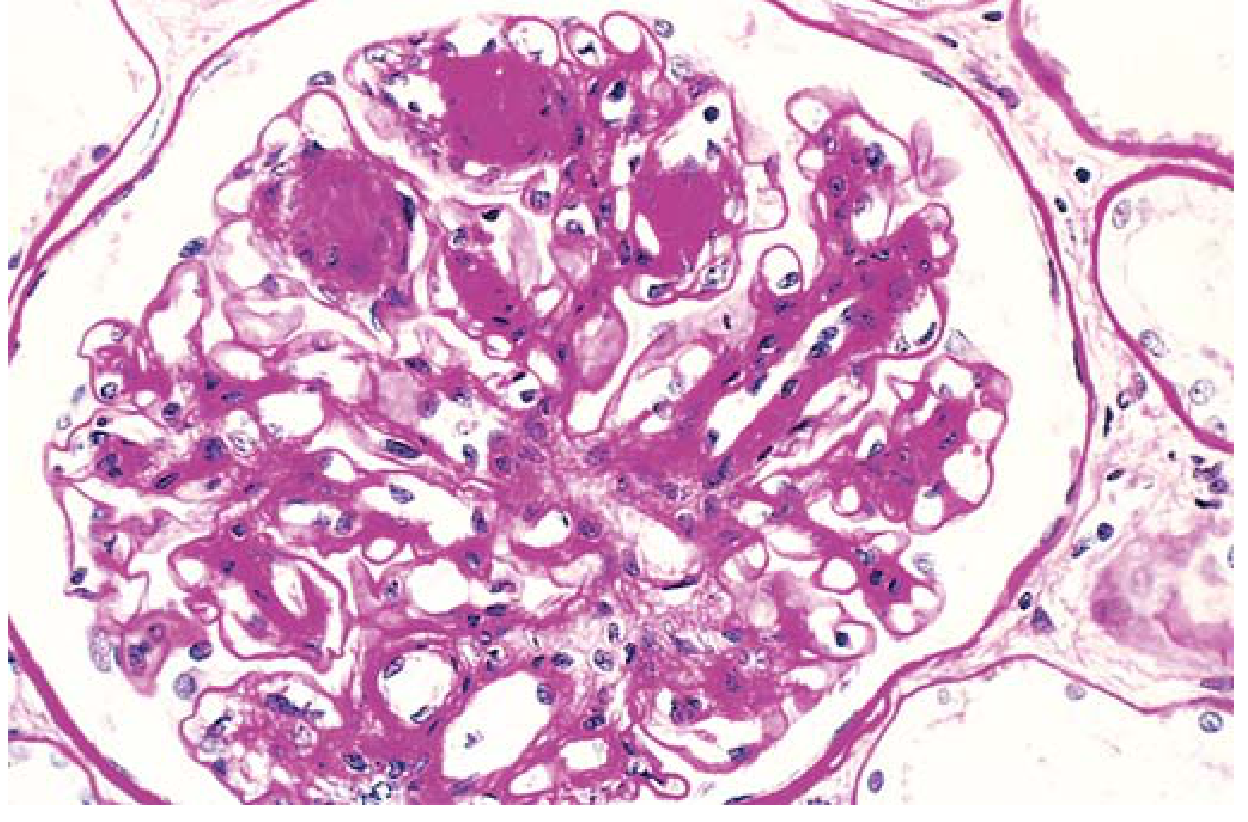
Eyes
- Diabetic retinopathy: Background (preproliferative) retinopathy — microaneurysms, dot-blot haemorrhages, hard exudates, cotton-wool spots; Proliferative retinopathy — neovascularisation driven by VEGF, vitreous haemorrhage, retinal detachment
- Cataracts: Sorbitol accumulation in the lens (polyol pathway) → osmotic injury → lens opacification
- Glaucoma: Increased intraocular pressure → optic nerve damage
Nervous System — Diabetic Neuropathy
- Peripheral symmetric polyneuropathy (most common) — predominantly sensory loss in stocking-glove distribution
- Autonomic neuropathy — gastroparesis, orthostatic hypotension, bladder dysfunction, impotence
- Mechanism: microangiopathy of vasa nervorum + direct axonal damage from sorbitol accumulation and AGEs
Infections
- Increased susceptibility to bacterial and fungal infections (impaired neutrophil function, poor tissue perfusion)
- Particularly: foot infections, urinary tract infections, mucormycosis, malignant otitis externa (Pseudomonas aeruginosa), "emphysematous" infections (gas-forming organisms)
Long-Term Complications Overview
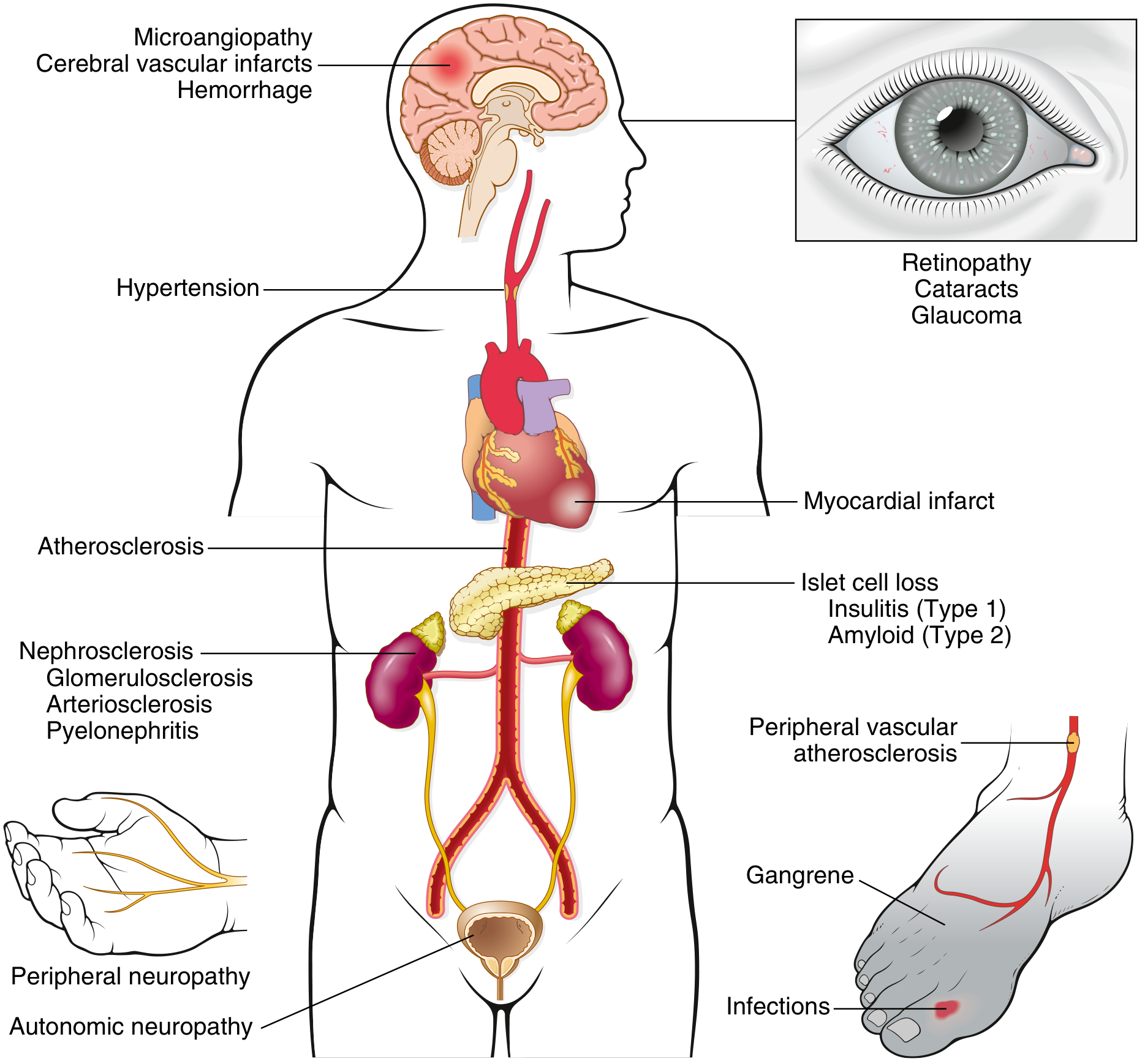
Acute Complications
| Complication | Mechanism | Type |
|---|---|---|
| DKA | Absolute insulin deficiency → lipolysis → ketogenesis | T1D primarily |
| HHS (Hyperosmolar Hyperglycemic State) | Extreme hyperglycemia (600–1200 mg/dL) + osmotic dehydration, no significant ketosis | T2D primarily |
| Hypoglycemia | Excess insulin dosing, missed meals, exercise | Both types on treatment |
Gestational Diabetes Mellitus (GDM)
- Glucose intolerance first detected during pregnancy (typically 2nd–3rd trimester)
- Caused by insulin resistance from placental hormones (human placental lactogen, progesterone, cortisol)
- Risks to fetus: macrosomia, neonatal hypoglycemia, respiratory distress syndrome
- ~50% of women with GDM develop T2D within 5–10 years postpartum
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (pp. 1014–1026); Robbins & Kumar Basic Pathology (pp. 743–752)
This is a shared conversation. Sign in to Orris to start your own chat.