A 40-year-old male presented with history of high-grade fever. Patient had weak rapid pulse, hypotension and tachypnea, cold clammy and cyanotic skin. Blood culture reveals Gram negative bacterial infection. What is your diagnosis ? Explain the pathogenesis and morphology with examples. Brief about other possible causes of your diagnosis.
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Searching PubMed
septic shock pathogenesis gram negative
I now have all the content needed for a comprehensive answer. Let me compose the full response.
Diagnosis: Septic Shock
The clinical triad of high-grade fever following a documented gram-negative bacteremia, combined with hypotension, weak rapid (thready) pulse, tachypnea, and cold/clammy/cyanotic skin represents septic shock - a subset of sepsis in which particularly profound circulatory, cellular, and metabolic abnormalities are associated with a greater risk of mortality than sepsis alone.
Searching sources...
Pathogenesis of Septic Shock
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, pp. 135-137)
Septic shock results from a dysregulated, generalized host response to infection that injures normal tissues remote from the initial site. The major pathogenic factors are as follows:
1. Inflammatory and Counter-inflammatory Responses
Gram-negative bacteria release LPS (lipopolysaccharide / endotoxin), which acts as the prime initiator. LPS and other microbial cell wall constituents (PAMPs - pathogen-associated molecular patterns) bind to Toll-like receptors (TLRs) on macrophages, neutrophils, dendritic cells, and endothelial cells.
- TLR ligation activates NF-kB, driving transcription of pro-inflammatory cytokines: TNF, IL-1, IL-12, IL-18, IFN-γ, and HMGB1
- Reactive oxygen species (ROS), prostaglandins (PGs), and platelet-activating factor (PAF) are elaborated
- Complement is activated - producing anaphylatoxins (C3a, C5a), chemotactic fragments (C5a), and opsonins (C3b)
- Acute phase reactants - CRP and procalcitonin - rise in the blood
Simultaneously, counter-regulatory immunosuppression develops - a shift from Th1 (pro-inflammatory) to Th2 (anti-inflammatory) cytokines, production of IL-10 and IL-1 receptor antagonist, lymphocyte apoptosis, and cellular anergy. This creates the characteristic oscillation between hyperinflammation and immune suppression.
2. Endothelial Activation and Injury
- Pro-inflammatory cytokines loosen endothelial tight junctions, causing widespread vascular leakage and protein-rich edema throughout the body
- Activated endothelium overproduces nitric oxide (NO) and other vasodilators (C3a, C5a, PAF), causing vascular smooth muscle relaxation and systemic hypotension
- Microvascular dysfunction develops: heterogeneous capillary flow, loss of autoregulation, mismatch between oxygen delivery and demand
3. Induction of a Procoagulant State (DIC)
This is a critical feature, occurring in up to 50% of septic patients:
- Pro-inflammatory cytokines increase tissue factor (TF) expression on monocytes and endothelium
- Anticoagulant factors are suppressed: TFPI (tissue factor pathway inhibitor), thrombomodulin, and Protein C are downregulated
- PAI-1 (plasminogen activator inhibitor-1) is increased, dampening fibrinolysis
- Neutrophil extracellular traps (NETs) promote both intrinsic and extrinsic coagulation pathways
- Result: Systemic thrombin activation → fibrin-rich microthrombi in small vessels → further ischemia
- Full-blown DIC causes consumption of clotting factors and platelets → simultaneous bleeding and clotting
4. Metabolic Abnormalities
- TNF and IL-1 + stress hormones (glucagon, growth hormone, glucocorticoids, catecholamines) drive gluconeogenesis → hyperglycemia
- Insulin release is suppressed; GLUT-4 expression is impaired → insulin resistance
- Hyperglycemia suppresses neutrophil bactericidal activity and upregulates endothelial adhesion molecules
- Lactic acidosis develops due to cellular hypoxia and impaired oxidative phosphorylation
- Adrenal insufficiency may supervene; in severe DIC, adrenal necrosis causes Waterhouse-Friderichsen syndrome
5. Organ Dysfunction - Stages of Shock
| Stage | Features |
|---|---|
| Nonprogressive | Compensatory: baroreceptors, catecholamines, ADH, renin-angiotensin → tachycardia, peripheral vasoconstriction, renal fluid conservation. Blood shunted to heart and brain |
| Progressive | Widespread tissue hypoxia → anaerobic glycolysis → lactic acidosis → arteriolar dilation → peripheral blood pooling → worsening cardiac output → DIC risk |
| Irreversible | Lysosomal enzyme leakage, myocardial depression by NO, intestinal flora translocation → bacteremic superinfection → renal failure, anuria, death |
Pathogenesis Diagram (Robbins)
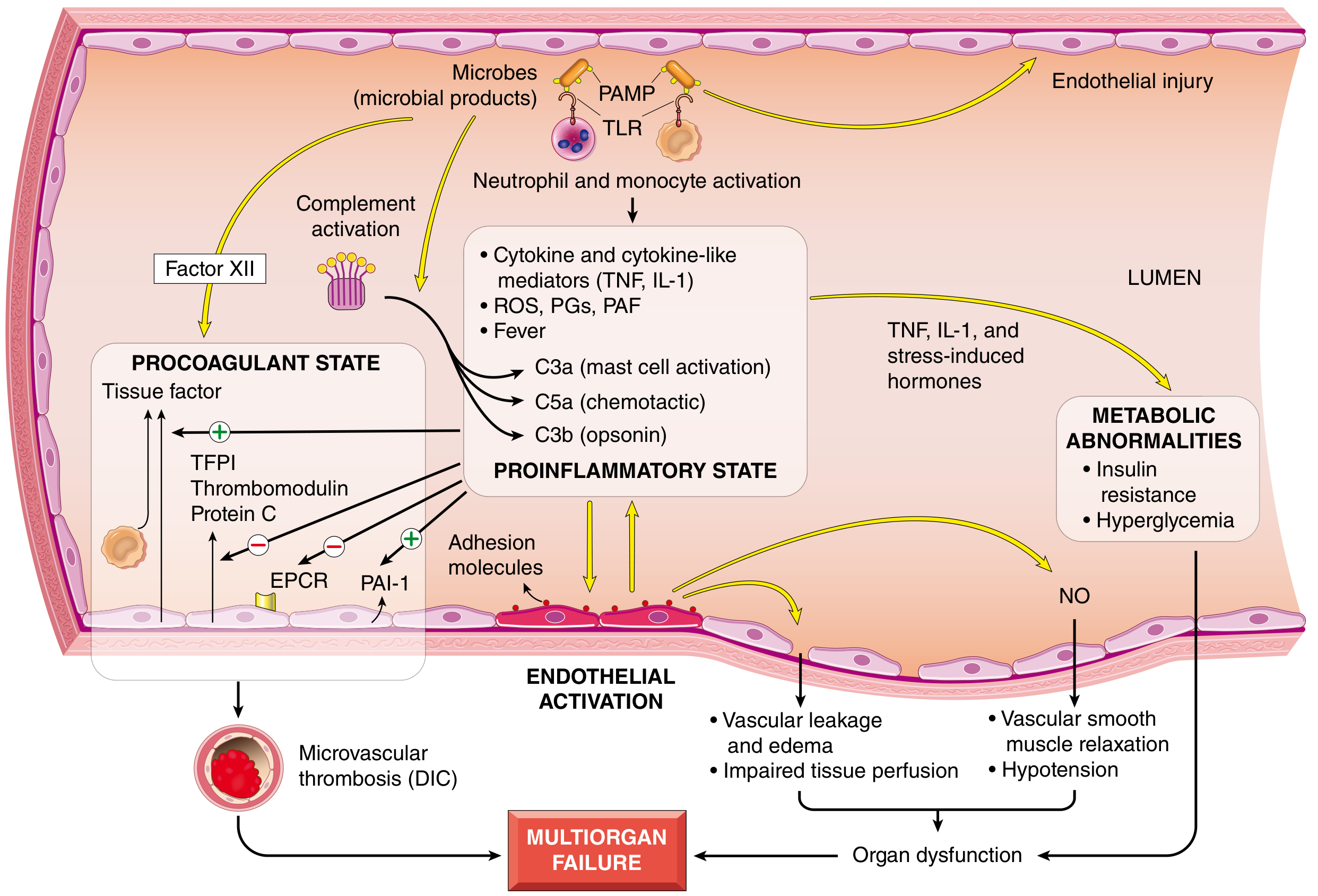
Fig. 4.20 from Robbins Pathology - Major pathogenic pathways in septic shock
Morphology of Septic Shock
(Robbins, Cotran & Kumar, p. 137)
The cellular changes are essentially those of hypoxic injury, caused by a combination of hypoperfusion and microvascular thrombosis. Organs most affected:
| Organ | Morphological Change |
|---|---|
| Kidney | Fibrin thrombi in glomerular capillaries (most prominent); acute tubular necrosis (ATN) from ischemia → oliguria progressing to anuria |
| Lung | In septic shock specifically: Diffuse alveolar damage (DAD) - "shock lung" / ARDS. Intra-alveolar hyaline membranes, edema, hemorrhage. Note: lungs are more resistant in pure hypovolemic shock |
| Brain | Neuronal ischemic injury; ischemic encephalopathy |
| Heart | Subendocardial hemorrhages; myocardial micronecrosis; cardiomyocyte loss |
| Adrenal glands | Cortical cell lipid depletion (stored lipids used for stress-related steroid synthesis); in DIC → Waterhouse-Friderichsen syndrome (bilateral adrenal hemorrhage and necrosis) |
| GI tract | Mucosal ischemia and hemorrhage; "stress ulcers" |
| Liver | Centrilobular necrosis; cholestasis |
Except for neuronal and cardiomyocyte loss (irreversible), affected tissues can recover completely if the patient survives.
Example of morphological finding: Fibrin thrombi in renal glomeruli are the most easily visualized on histology, representing the DIC component of septic shock. In "shock lung" (ARDS), hyaline membranes lining alveolar walls are a hallmark finding on histopathology.
Other Causes of Shock (Classification)
Shock is broadly classified into the following types, each with distinct causes:
1. Cardiogenic Shock
Failure of the heart as a pump:
- Myocardial infarction (most common cause)
- Myocarditis
- Cardiac tamponade
- Severe arrhythmias
- Dilated cardiomyopathy
- Massive pulmonary embolism
2. Hypovolemic Shock
Reduced circulating blood volume:
- Hemorrhagic: trauma, ruptured aortic aneurysm, GI bleeding, obstetric hemorrhage
- Non-hemorrhagic: severe burns (plasma loss), vomiting/diarrhea, Addison's disease (salt-wasting)
3. Septic Shock (Distributive)
Systemic infection with dysregulated vasodilation:
- Gram-negative bacteria (current case): E. coli, Klebsiella, Pseudomonas, Neisseria meningitidis, Proteus - via endotoxin (LPS)
- Gram-positive bacteria (most common overall): Staphylococcus aureus, Streptococcus pyogenes - via superantigens (toxic shock syndrome), teichoic acid
- Fungi: Candida (especially in immunocompromised)
- Viruses: SARS-CoV-2, influenza (emerging causes)
- Anaerobes: Clostridium, bacteroides
4. Anaphylactic Shock (Distributive)
IgE-mediated type I hypersensitivity:
- Drugs (penicillin, NSAIDs), bee stings, food allergens (peanuts)
- Histamine release → venodilation → reduced venous return → severe hypotension
5. Neurogenic Shock (Distributive)
Loss of vasomotor tone:
- Spinal cord injury (above T6)
- Deep general anesthesia
- Brain injury
6. Obstructive Shock
Physical obstruction to blood flow:
- Massive pulmonary embolism
- Tension pneumothorax
- Cardiac tamponade
Key Summary
| Feature | This Case |
|---|---|
| Diagnosis | Septic Shock |
| Causative agent | Gram-negative bacteria (LPS/endotoxin) |
| Primary trigger | LPS → TLR4 activation → NF-kB → cytokine storm |
| Key mediators | TNF, IL-1, NO, PAF, ROS, complement |
| Hemodynamic pattern | Distributive - initially high CO, low SVR |
| Coagulation | DIC in up to 50% |
| Morphology | ATN (kidney), DAD/ARDS (lung), adrenal lipid depletion, cerebral ischemia, GI ulcers |
| Mortality | ~40% despite modern care |
References: Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.), pp. 134-138; Guyton and Hall - Textbook of Medical Physiology, p. 2369.
25 years male RTA victim presented with rapid pulse, cold clammy cyanotic skin, tachypnea and landed in emergency ward in unconscious state what is the diagnosis ? Classify the disease write in detail about the pathogenesis, morphology evolution of the disease.
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I have all the content I need - comprehensive data from Guyton (physiology/pathogenesis), Mulholland & Greenfield Surgery (ATLS classification), Costanzo Physiology (case-based explanation), and Robbins (morphology). Let me now compose the full answer.
Diagnosis: Hypovolemic (Hemorrhagic) Shock
A 25-year-old male RTA (road traffic accident) victim presenting with rapid thready pulse, cold/clammy/cyanotic skin, tachypnea, and unconsciousness has suffered massive acute blood loss. This is classic hypovolemic shock due to hemorrhage, the most common form of shock encountered in trauma/surgical practice.
Classification of Shock
Shock is a state of systemic tissue hypoperfusion due to reduced cardiac output and/or reduced effective circulating volume. The broad classification:
| Type | Subtype | Common Causes |
|---|---|---|
| Hypovolemic | Hemorrhagic | Trauma (RTA), GI bleeding, ruptured aneurysm |
| Non-hemorrhagic | Burns (plasma loss), vomiting/diarrhea, third-spacing | |
| Cardiogenic | Myocardial | MI, cardiomyopathy, myocarditis, contusion |
| Valvular/Arrhythmic | Ruptured papillary muscle, VT/VF | |
| Obstructive | Extracardiac | Tension pneumothorax, cardiac tamponade, massive PE |
| Distributive | Septic | Gram-negative/positive bacteria, fungi |
| Anaphylactic | Drugs, insect venom | |
| Neurogenic | Spinal cord injury, deep anesthesia |
(Mulholland and Greenfield's Surgery, p. 532-534)
ATLS Classification of Hemorrhagic Shock (American College of Surgeons)
Estimated total blood volume = ~4,900 mL in a 70-kg patient (7% of ideal body weight)
| Class | Blood Loss | Clinical Features | Treatment |
|---|---|---|---|
| I | Up to 15% (<750 mL) | No measurable change in HR, BP, or RR. Normal capillary refill | Minimal - self-corrects |
| II | 15-30% (800-1,500 mL) | Tachycardia, tachypnea, narrow pulse pressure, anxiety, slightly decreased urine output (20-30 mL/hr), delayed capillary refill | Crystalloids ± blood |
| III | 30-40% (up to 2,000 mL) | Marked tachycardia and tachypnea, hypotension, cold/clammy extremities, confusion/combativeness, significant oliguria | Blood transfusion + crystalloids |
| IV | >40% (>2,000 mL) | Marked tachycardia, severely depressed BP, obtunded/comatose, cold pale skin, negligible urine output - LIFE-THREATENING | Immediate transfusion + surgical intervention |
This patient - unconscious, cold/clammy/cyanotic, rapid pulse, hypotension, tachypnea = Class III-IV hemorrhagic shock (>30-40% blood volume loss)
Pathogenesis of Hypovolemic Shock
(Guyton & Hall Textbook of Medical Physiology; Costanzo Physiology 7th Ed.)
Initial Trigger
Massive hemorrhage from trauma → reduced circulating blood volume → reduced venous return → reduced cardiac filling → fall in cardiac output → fall in arterial pressure.
About 10% blood volume loss causes no measurable change. When 40-45% is lost, both cardiac output and arterial pressure fall to zero.
Stage 1: Compensatory (Nonprogressive) Phase
The body immediately activates multiple compensatory mechanisms:
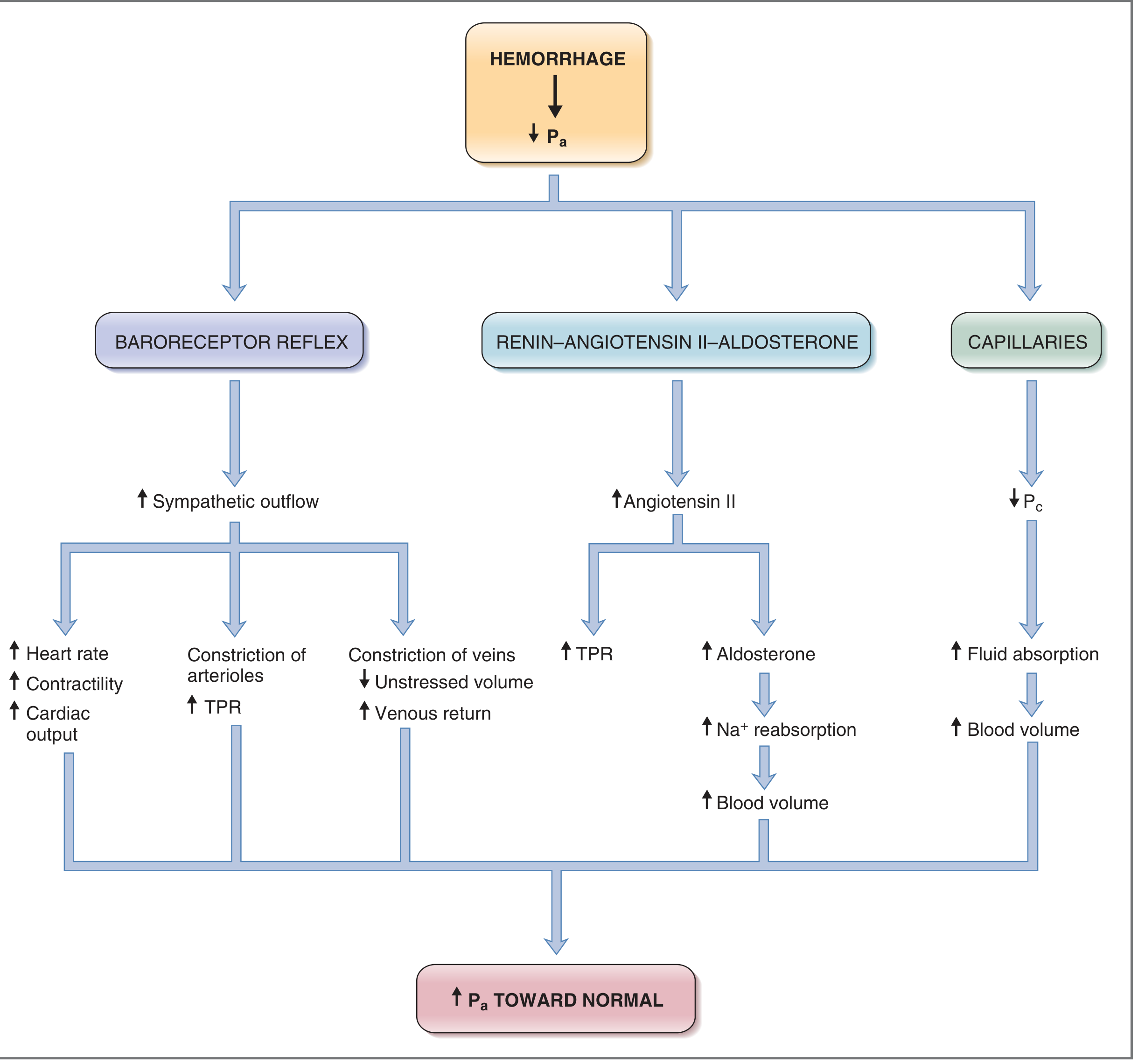
A. Baroreceptor Reflex (rapid, within seconds)
- Arterial baroreceptors in carotid sinus and aortic arch detect the fall in BP
- Increased sympathetic outflow → tachycardia, increased myocardial contractility
- Arteriolar vasoconstriction (except coronary and cerebral vessels) → increased total peripheral resistance (TPR)
- Venous constriction → increased venous return to heart
- Result: Blood shunted from skin, gut, and skeletal muscle to vital organs (heart and brain)
- Cutaneous vasoconstriction explains the cold, pale, clammy skin
B. Renin-Angiotensin-Aldosterone System (RAAS) (10-60 min)
- Reduced renal perfusion → renin release → Angiotensin II → arteriolar vasoconstriction + aldosterone release → Na⁺ and water retention
C. ADH / Vasopressin (from posterior pituitary)
- Released in response to decreased blood volume and osmolality → vasoconstriction + renal water conservation
D. Adrenal Medullary Response
- Release of epinephrine and norepinephrine → peripheral vasoconstriction, tachycardia, increased cardiac contractility
E. Capillary Fluid Shift (hours)
- Reduced capillary hydrostatic pressure → net interstitial fluid absorption into capillaries → auto-transfusion effect, partially restoring blood volume
F. CNS Ischemic Response
- When BP falls below 50 mmHg, cerebral ischemia itself triggers extreme sympathetic discharge ("last ditch stand"), producing a temporary secondary plateau in arterial pressure at ~50 mmHg
Cardiovascular Compensation Diagram (Costanzo Physiology)
Stage 2: Progressive Phase - The Vicious Cycle
If hemorrhage exceeds a critical threshold, shock becomes self-perpetuating through multiple positive feedback loops:
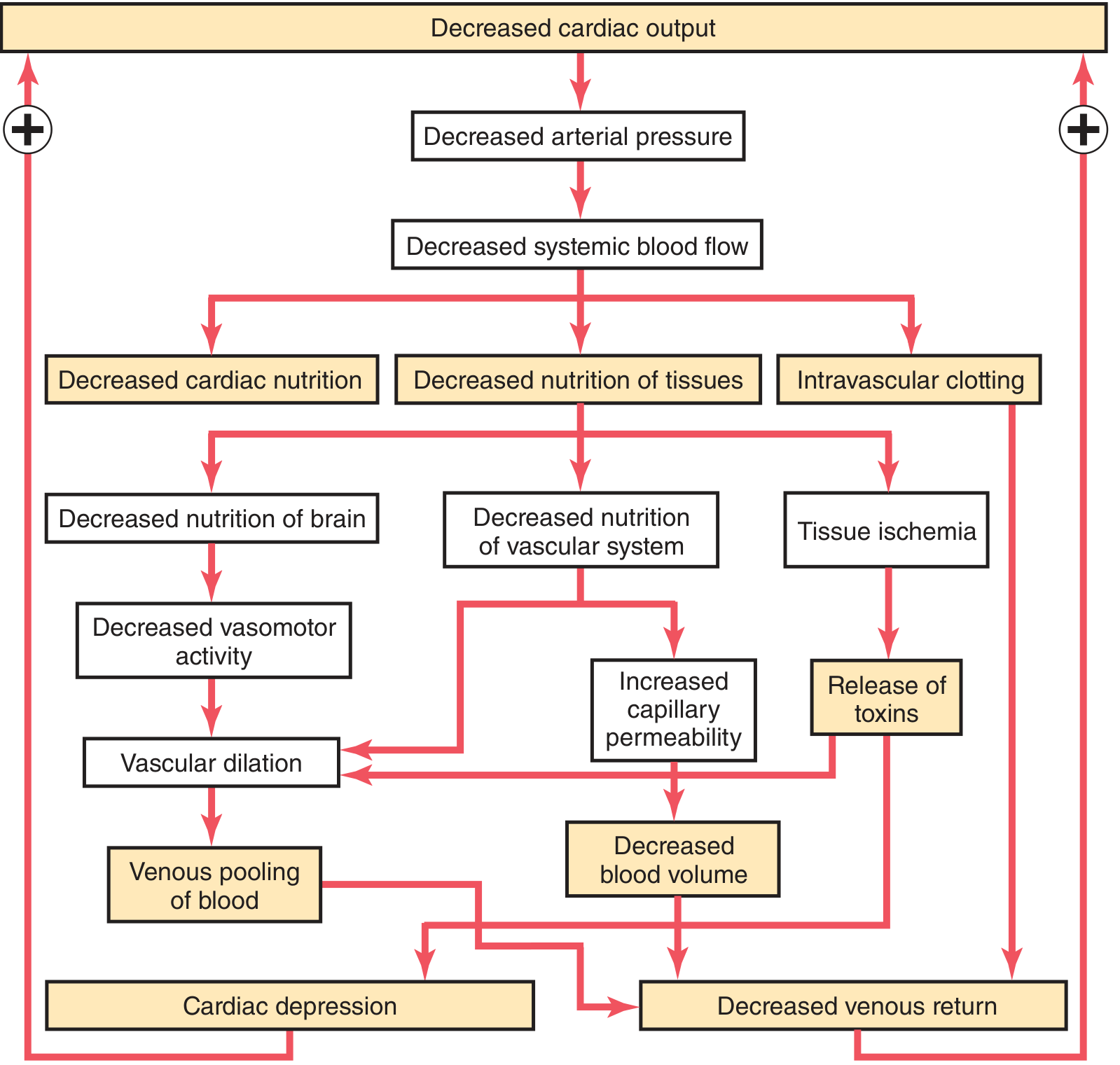
Key vicious cycles:
-
Cardiac depression: Prolonged low coronary blood flow → myocardial ischemia → further fall in cardiac output → more ischemia. By 4 hours at arterial pressure of 30 mmHg, cardiac output falls ~40%; then rapidly to zero.
-
Vasomotor centre failure: Prolonged cerebral hypoperfusion → vasomotor centre in medulla depresses → arteriolar dilation → more venous pooling → further fall in cardiac output
-
Microvascular sludging and thrombosis:
- Tissue hypoxia + lactic/carbonic acid accumulation → local blood agglutination → small vessel plugging
- Intravascular clotting → further impairs microvascular flow
- Increased capillary permeability:
- Prolonged capillary ischemia damages the capillary walls → protein-rich fluid leaks into interstitium → blood volume further falls
- Release of inflammatory toxins:
- Ischemic tissues release histamine, serotonin, cytokines, and lysosomal enzymes → further vascular damage
- In prolonged shock: intestinal ischemia → bacterial translocation → endotoxin absorption → cardiac depression (superimposed endotoxemia)
- Generalized cellular deterioration:
- Failure of Na⁺/K⁺-ATPase → cellular sodium and water accumulation, cellular swelling
- Mitochondrial dysfunction → impaired oxidative phosphorylation → lactic acidosis
- Lysosomal rupture → intracellular release of hydrolases → cell death
- Insulin resistance and glucose utilization failure
Stage 3: Irreversible Phase
- Widespread cell death reaches a point where even if hemodynamics are restored, the patient cannot survive
- Characterized by: anuria, profound metabolic acidosis, refractory hypotension, multiorgan failure
- The conscious patient from Stage 2 (confusion, combativeness) progresses to unconsciousness - as seen in this case - indicating Stage 3 involvement
Morphology of Hypovolemic Shock
(Robbins, Cotran & Kumar - Pathologic Basis of Disease, p. 137)
The cellular changes are those of ischemic/hypoxic injury from hypoperfusion and microvascular thrombosis. Any organ can be affected, but the following are most prominent:
Brain
- Ischemic encephalopathy - neuronal cell death in areas most vulnerable to hypoxia (hippocampus CA1, Purkinje cells of cerebellum, layers 3, 5, 6 of cortex)
- This explains the unconscious state in this patient
- Neuronal loss is irreversible
Heart
- Subendocardial hemorrhage and coagulative necrosis (myocardial infarction-like changes in subendocardial zones)
- These areas are most remote from blood supply and most vulnerable
- Cardiomyocyte loss is irreversible
Kidney
- Acute Tubular Necrosis (ATN) - the most common renal manifestation
- Proximal tubular and ascending loop of Henle cells are most susceptible
- Histology: dilated tubular lumens with flattened epithelium, loss of brush border, sloughed epithelial cells as granular casts in tubules
- Fibrin thrombi may be seen in glomerular capillaries
- Clinically → oliguria → anuria
Lung
- In pure hypovolemic shock: relatively resistant (well protected by collateral circulation)
- In trauma-associated shock: Diffuse Alveolar Damage (DAD) / ARDS ("Shock Lung")
- Precipitated by the combination of trauma and hypoperfusion
- Histology: hyaline membranes, interstitial edema, hemorrhage, type II pneumocyte hyperplasia
Liver
- Centrilobular (zone 3) necrosis - as this zone is furthest from blood supply (perivenular)
- Illustrated by Guyton's Fig. 24.5: patchy necrosis of central portions of hepatic lobules near central veins
- Hepatic ischemia → reduced metabolic and detoxification function → worsens overall metabolic derangements
GI Tract
- Mucosal ischemia → hemorrhagic erosions (stress ulcers), primarily in stomach and duodenum
- Intestinal barrier failure → bacterial/endotoxin translocation → superimposed sepsis/endotoxemia
Adrenal Glands
- Cortical cell lipid depletion - stored cholesterol esters used for emergency cortisol synthesis
- In severe DIC: bilateral adrenal hemorrhage (Waterhouse-Friderichsen syndrome)
Evolution Summary
RTA → Hemorrhage
↓
Reduced Blood Volume (>30-40%)
↓
Stage 1: Compensatory (Nonprogressive)
• Baroreceptor reflex → tachycardia, vasoconstriction
• RAAS → fluid retention
• ADH → fluid conservation
• Adrenal catecholamines → tachycardia, vasoconstriction
[Clinical: Rapid pulse, clammy skin, tachypnea - CONSCIOUS]
↓ (if untreated / massive hemorrhage)
Stage 2: Progressive (Vicious Cycle Begins)
• Cardiac depression, vasomotor failure
• Microvascular sludging/DIC
• Capillary leak, cellular deterioration
• Lactic acidosis, cytokine storm
[Clinical: Confusion, worsening hypotension]
↓
Stage 3: Irreversible
• Widespread cell death (neurons, cardiomyocytes)
• Multiorgan failure (ATN, DAD, hepatic necrosis)
• Refractory metabolic acidosis
[Clinical: UNCONSCIOUS, no urine - as in this patient]
Key Hemodynamic Profile of Hypovolemic Shock
| Parameter | Change |
|---|---|
| Cardiac Output (CO) | ↓↓ |
| Systemic Vascular Resistance (SVR) | ↑↑ |
| Pulmonary Artery Occlusion Pressure (PAOP) | ↓ |
| Central Venous Pressure (CVP) | ↓ |
| Mixed venous O₂ saturation (SvO₂) | ↓ |
(Mulholland and Greenfield's Surgery, Table 9.3)
This profile distinguishes hypovolemic shock from distributive shock (where SVR is low) and cardiogenic shock (where PAOP is elevated).
References: Guyton & Hall - Textbook of Medical Physiology (14th ed.), pp. 299-305; Mulholland & Greenfield's Surgery (7th ed.), pp. 530-535; Costanzo - Physiology (7th ed.), p. 187; Robbins, Cotran & Kumar - Pathologic Basis of Disease (10th ed.), pp. 136-137.
This is a shared conversation. Sign in to Orris to start your own chat.