
IMG_20260621_103621_330.jpg
Explain whole diabetes
Reading File
Finding Sources
Finding Sources
Searching PubMed
"diabetes mellitus"[MeSH Terms] AND "pathophysiology"[MeSH Terms]
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have all the content needed for a thorough answer. Let me compile the complete explanation.
Diabetes Mellitus - Complete Explanation
Source: Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.), Chapter 24 - The Endocrine System
1. Definition
Diabetes mellitus is a group of metabolic disorders sharing the common feature of hyperglycemia caused by defects in insulin secretion, insulin action, or (most commonly) both. The chronic hyperglycemia and attendant metabolic deregulation cause secondary damage in multiple organ systems, especially the kidneys, eyes, nerves, and blood vessels.
In the United States, diabetes is:
- The leading cause of end-stage renal disease
- The leading cause of adult-onset blindness
- The leading cause of nontraumatic lower extremity amputations
2. Epidemiology
- Affects >30 million people (>11% of the US population)
- ~1.9 million have Type 1 diabetes (T1D); the rest have Type 2 (T2D)
- ~96 million US adults have "prediabetes" (impaired glucose tolerance not yet meeting the T2D threshold)
- Worldwide: 422 million people affected (WHO estimate)
- T2D rates in adolescents have increased dramatically in recent years
- Native Americans, African Americans, and Hispanics are 1.5-2x more likely to develop diabetes
- 7th leading cause of death in the US (2019); total yearly cost ~$327 billion
3. Diagnosis (ADA/WHO Criteria)
Normal fasting blood glucose = 70-120 mg/dL
A diagnosis of diabetes requires any ONE of the following:
| Criterion | Threshold |
|---|---|
| Fasting plasma glucose | ≥ 126 mg/dL |
| Random plasma glucose (with symptoms) | ≥ 200 mg/dL |
| 2-hour glucose during 75g OGTT | ≥ 200 mg/dL |
| HbA1c | ≥ 6.5% |
Prediabetes = impaired fasting glucose (100-125 mg/dL) or HbA1c 5.7-6.4%
HbA1c (glycated hemoglobin) reflects average blood glucose over the preceding 2-3 months and is used to monitor long-term glycemic control.
4. Classification
| Category | Description |
|---|---|
| Type 1 (T1D) | Autoimmune destruction of β cells |
| Type 2 (T2D) | Insulin resistance + β-cell failure |
| Gestational DM | New-onset during pregnancy |
| Monogenic (MODY) | Single-gene defects in β-cell function |
| Secondary DM | Due to drugs, other diseases (pancreatitis, Cushing's, etc.) |
5. Normal Insulin Physiology (Basis for Understanding Diabetes)
Insulin is produced in pancreatic β cells as a precursor, cleaved in the Golgi into mature insulin + C-peptide (both secreted in equal amounts - C-peptide is used as a marker of β-cell function).
Glucose-stimulated insulin release:
- Glucose enters the β cell via GLUT-2 transporter
- Glucokinase phosphorylates glucose → glucose-6-phosphate (rate-limiting step)
- Increased ATP:ADP ratio closes K⁺-ATP channels → membrane depolarization
- Voltage-gated Ca²⁺ channels open → insulin granule exocytosis
Insulin actions on target tissues:
- Liver: inhibits gluconeogenesis and glycogenolysis; promotes glycogen synthesis
- Skeletal muscle: stimulates glucose uptake (via GLUT-4 translocation) and glycogen synthesis
- Adipose tissue: promotes lipogenesis; inhibits lipolysis
6. TYPE 1 DIABETES (T1D)
Pathogenesis
T1D is an autoimmune disease caused by selective destruction of pancreatic β cells by autoreactive T lymphocytes.
Genetic susceptibility:
- Major linkage to MHC class II genes (HLA-DR3, HLA-DR4; especially DR3/DR4 heterozygotes have highest risk)
- Polymorphisms in CTLA4 and PTPN22 (both regulate T-cell activity)
- Insulin gene VNTR polymorphisms affect thymic insulin expression
Environmental factors:
- Concordance in identical twins is only ~50%, so environment is critical
- Viral infections (e.g., enteroviruses, Coxsackie B) may trigger autoimmunity via molecular mimicry (shared epitopes between viral proteins and islet antigens)
- Some infections are thought to be protective against T1D (hygiene hypothesis)
Three Stages of T1D
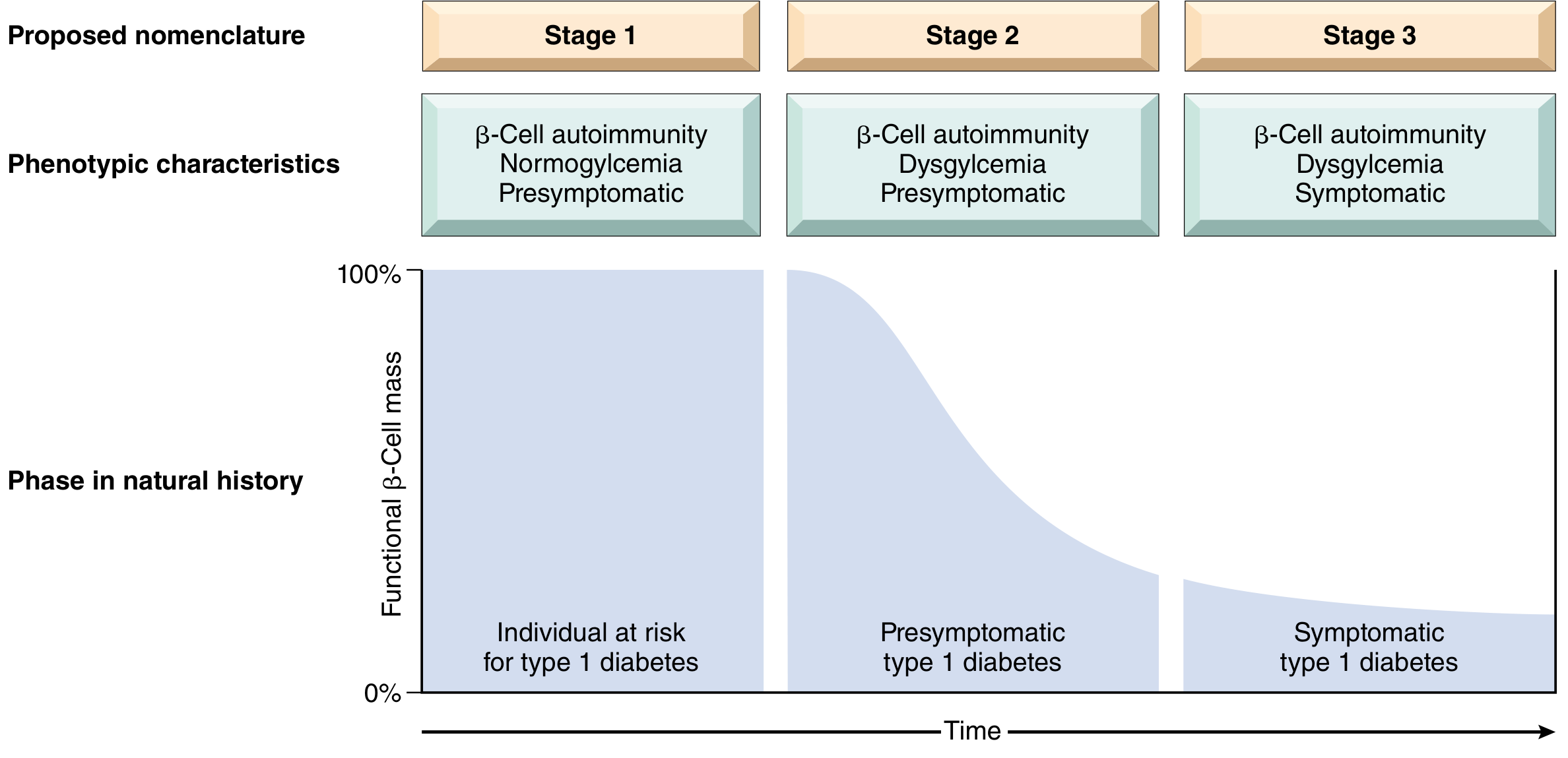
| Stage | Features | 5-year Risk of Symptomatic T1D |
|---|---|---|
| Stage 1 | ≥2 islet autoantibodies, normoglycemia, presymptomatic | <50% |
| Stage 2 | Autoantibodies + dysglycemia (glucose tolerance impairment), presymptomatic | 75% |
| Stage 3 | Symptomatic (polyuria, polydipsia, DKA) - >90% β-cells destroyed | 100% |
Mechanism of β-Cell Destruction
- Autoreactive T cells that escape clonal deletion in the thymus are activated in peripancreatic lymph nodes
- Th1 CD4+ cells secrete IFN-γ and TNF, causing β-cell injury
- CD8+ cytotoxic T cells directly kill β cells
- Target antigens include: insulin, glutamic acid decarboxylase (GAD65), IA-2, ZnT8
- Autoantibodies (anti-GAD65, anti-IA2, anti-ZnT8) are found at presymptomatic stages - useful for screening
- Insulitis: lymphocytic infiltration of islets (mainly T cells and macrophages)
Clinical Presentation of T1D
- Onset typically in childhood/adolescence (but can occur at any age)
- Patients are usually normal weight or underweight
- Abrupt onset of: polyuria, polydipsia, polyphagia, weight loss
- "Honeymoon period" in first 1-2 years - residual β-cell function transiently maintains some insulin production
- Diabetic ketoacidosis (DKA) is the hallmark acute complication
- Requires insulin therapy for life
7. TYPE 2 DIABETES (T2D)
Pathogenesis
T2D involves two key abnormalities:
1. Insulin Resistance - decreased response of peripheral tissues (skeletal muscle, adipose, liver) to insulin
2. β-Cell Dysfunction - inadequate insulin secretion in the face of resistance and hyperglycemia
In early T2D, insulin resistance is compensated by β-cell hyperfunction and hyperinsulinemia. Over years, β cells "burn out," secretory capacity falls, and frank hyperglycemia develops.
Insulin Resistance
In insulin-resistant states:
- Liver: fails to suppress gluconeogenesis → high fasting blood glucose
- Skeletal muscle: fails to uptake glucose post-meal → high postprandial glucose
- Adipose tissue: fails to suppress lipases → excess free fatty acids (FFAs) in circulation
Mechanistically: reduced tyrosine phosphorylation of insulin receptor substrate (IRS) proteins → reduced GLUT-4 translocation to cell surface in muscle
Role of Obesity
Obesity (especially central/visceral adiposity) is the most important cause of insulin resistance. BMI correlates with T2D risk.
How obesity causes insulin resistance:
- Excess FFAs overwhelm β-oxidation pathways → accumulation of diacylglycerol (DAG), ceramides - these "toxic lipids" inhibit insulin receptor signaling and activate inflammatory pathways in islets
- Adipokines: Adipose tissue in obese individuals secretes more resistin and TNF-α (pro-insulin resistance) and less adiponectin (which normally enhances insulin sensitivity)
- Chronic low-grade inflammation: macrophage infiltration of visceral fat releases IL-6, TNF-α → systemic insulin resistance
β-Cell Failure in T2D
- Genetic factors (~40% heritability), linked to TCF7L2, PPARG, FTO polymorphisms (not HLA)
- Chronic exposure to excess glucose (glucotoxicity) and FFAs (lipotoxicity) impairs β-cell function
- Amyloid deposition (islet amyloid polypeptide/IAPP) in islets - obliterates islet architecture in advanced T2D
Clinical Presentation of T2D
- Onset typically in adults (increasingly in adolescents)
- 80% are obese at diagnosis
- Often insidious and asymptomatic - detected incidentally on routine labs
- May present with complications (neuropathy, retinopathy) already established
- Nonketotic hyperosmolar coma (rather than DKA) is the characteristic acute crisis
- Circulating insulin is initially elevated (early) then normal or decreased (late)
- No islet autoantibodies
8. Comparison: Type 1 vs Type 2
| Feature | Type 1 | Type 2 |
|---|---|---|
| Age of onset | Usually childhood/adolescence | Usually adult (increasing in youth) |
| Body habitus | Normal weight / weight loss | 80% obese |
| Insulin levels | Progressive decrease | Increased (early), then normal/decreased |
| Autoantibodies | Yes (anti-GAD65, anti-IA2, anti-ZnT8) | No |
| HLA linkage | Yes (MHC class II) | No |
| Genetics | CTLA4, PTPN22, insulin gene VNTR | TCF7L2, PPARG, FTO |
| Pathology | Insulitis (T cells + macrophages) | Islet amyloid deposition |
| Acute complication | DKA | Nonketotic hyperosmolar coma |
| Treatment | Insulin (mandatory) | Diet/exercise, oral agents, insulin |
9. Monogenic Forms (MODY)
Maturity-Onset Diabetes of the Young (MODY) = germline loss-of-function mutations in single genes (~1-2% of diabetics):
- Glucokinase (GCK): Altered glucose "set point"; hyperglycemia but no typical complications
- HNF1α, HNF1β, HNF4α, PDX1 (various transcription factor mutations): cause β-cell dysfunction with typical complications
- Insulin gene mutations: directly reduce insulin production
Insulin receptor mutations (Type A insulin resistance): severe resistance with hyperinsulinemia, acanthosis nigricans, polycystic ovaries in females.
10. Pathophysiology of Complications (Biochemical Mechanisms)
Four major pathways link hyperglycemia to organ damage:
A. Advanced Glycation End Products (AGEs)
- Glucose non-enzymatically binds to proteins/lipids → AGEs
- AGEs cross-link collagen → thickened basement membranes (key in microangiopathy)
- AGEs bind RAGE receptors → activate NF-κB → inflammation and cytokine production
- AGEs trap plasma proteins in vessel walls
B. Polyol Pathway / Oxidative Stress
- Excess intracellular glucose → converted by aldose reductase to sorbitol (using NADPH as cofactor)
- NADPH also needed to regenerate reduced glutathione (GSH) - a key antioxidant
- Sorbitol accumulation depletes NADPH → less GSH → increased oxidative stress
- Sorbitol in the lens → cataracts
C. PKC Activation
- Hyperglycemia increases diacylglycerol (DAG) → activates Protein Kinase C (PKC)
- PKC signaling alters gene expression: increases VEGF (→ neovascularization), TGF-β (→ basement membrane thickening), endothelin-1 (→ vasoconstriction), and decreases eNOS (→ vasodilation)
D. Hexosamine Pathway
- Glycolytic intermediates shunted through hexosamine pathway → enhanced oxidative stress → insulin resistance and vascular complications
11. CHRONIC COMPLICATIONS
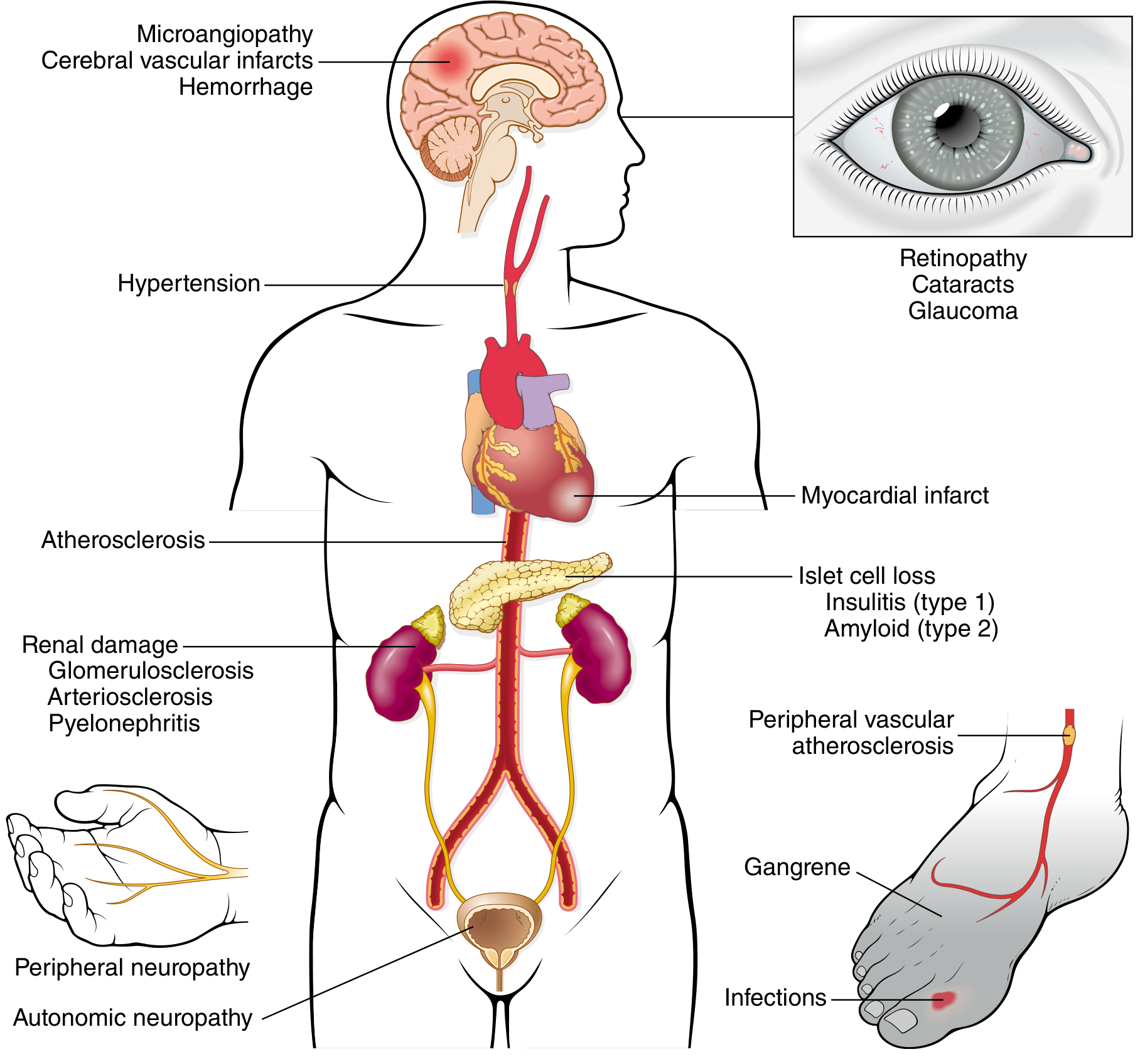
Chronic complications typically appear 15-20 years after onset. Their severity correlates with both the degree and duration of hyperglycemia.
A. Macrovascular Disease (Large Vessels)
- Accelerated atherosclerosis of aorta, coronary, and peripheral arteries
- Diabetics have 2-4x higher risk of coronary artery disease
- 4-fold higher risk of dying from cardiovascular causes
- Risk elevated even in prediabetes
- Hypertension is present in ~75% of T2D patients
- Diabetic dyslipidemia: increased triglycerides and LDL, decreased HDL
B. Diabetic Nephropathy
- Leading cause of end-stage renal disease (ESRD) in the USA
- 30-40% of all diabetics develop clinical nephropathy
- Progression:
- Microalbuminuria (30-300 mg/day) = earliest sign
- Overt nephropathy (macroalbuminuria >300 mg/day) + hypertension
- ESRD requiring dialysis/transplant
- Pathology: Kimmelstiel-Wilson nodules (nodular glomerulosclerosis), diffuse glomerulosclerosis, arteriosclerosis, pyelonephritis
C. Diabetic Retinopathy
- Affects 60-80% of patients - leading cause of adult blindness in USA
- Fundamental lesion: neovascularization driven by hypoxia-induced VEGF
- Treatment: anti-VEGF agents (e.g., bevacizumab, ranibizumab)
- Also: cataracts (sorbitol accumulation in lens), glaucoma
D. Diabetic Neuropathy
- Most common form: distal symmetric polyneuropathy of lower extremities (sensorimotor)
- "Glove-and-stocking" pattern when upper extremities involved
- Autonomic neuropathy: bowel/bladder dysfunction, erectile dysfunction, orthostatic hypotension
- Mononeuropathy: sudden footdrop, wrist-drop, cranial nerve palsies
E. Susceptibility to Infections
- Impaired neutrophil function (chemotaxis, phagocytosis) due to hyperglycemia
- Poor wound healing
- Predisposed to: mucormycosis, Candida, recurrent urinary tract infections, foot ulcers, gas-forming infections
12. ACUTE METABOLIC COMPLICATIONS
A. Diabetic Ketoacidosis (DKA) - primarily T1D
- Precipitated by insulin deficiency (+ stress, infection)
- Insulin lack → unrestrained lipolysis → excess FFAs → liver β-oxidation → ketone bodies (acetoacetate, β-hydroxybutyrate)
- Presents with: nausea, vomiting, abdominal pain, fruity breath, Kussmaul breathing, altered consciousness
- Labs: hyperglycemia, ketonemia, metabolic acidosis (anion gap), hypokalemia (after treatment)
- Treatment: IV fluids, insulin infusion, potassium replacement
B. Hyperosmolar Hyperglycemic State (HHS) - primarily T2D
- Extreme hyperglycemia (often >600 mg/dL) with severe dehydration
- No significant ketosis (residual insulin prevents ketogenesis)
- Presents with: profound dehydration, neurologic symptoms, coma
- Very high mortality if untreated
C. Hypoglycemia
- Most common acute complication in treated diabetics
- Causes: excess insulin/sulfonylurea, skipped meals, exercise
- Symptoms: sweating, tremor, palpitations (adrenergic), then confusion, seizures (neuroglycopenic)
- Treatment: glucose (oral or IV dextrose/glucagon if severe)
13. Morphological Changes in the Pancreas
- T1D: dramatic reduction in islet number and size; insulitis (lymphocytic infiltration); β-cell depletion
- T2D: subtle reduction in islet cell mass; amyloid deposition (IAPP) obliterating islets; no insulitis
- Nondiabetic newborns of diabetic mothers: islet hyperplasia (response to maternal hyperglycemia)
14. Diabetes and Pregnancy
- Gestational diabetes: new-onset during pregnancy (~5-9% of US pregnancies); pregnancy is "diabetogenic" due to placental hormones creating insulin resistance
- Pregestational diabetes: pre-existing diabetes complicating pregnancy - increased risk of congenital malformations and stillbirth
- Poorly controlled late-pregnancy diabetes → macrosomia (large newborn), neonatal hypoglycemia
- Gestational diabetes usually resolves postpartum but most affected women develop T2D within 10-20 years
Summary Chart
DIABETES MELLITUS
├── Type 1 (Autoimmune)
│ ├── HLA-linked → autoreactive T cells → insulitis → β-cell destruction
│ ├── Acute: DKA
│ └── Treatment: Insulin
│
├── Type 2 (Metabolic)
│ ├── Obesity → insulin resistance → β-cell compensation → β-cell failure
│ ├── Acute: Hyperosmolar nonketotic coma
│ └── Treatment: lifestyle, metformin, GLP-1 agonists, SGLT-2i, insulin
│
├── Monogenic (MODY, type A resistance)
├── Gestational
│
└── Chronic Complications (all types, 15-20 yrs)
├── Macrovascular: MI, stroke, peripheral vascular disease
├── Microvascular: Nephropathy, Retinopathy, Neuropathy
└── Infections
- Robbins, Cotran & Kumar Pathologic Basis of Disease, pp. 997-1010
This is a shared conversation. Sign in to Orris to start your own chat.