Types of jaunildice and bilirubin metabolism
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have all the information needed for a thorough, well-illustrated answer. Let me compile everything.
Jaundice: Types and Bilirubin Metabolism
Jaundice (icterus) is yellow discoloration of the skin, sclerae, and mucous membranes caused by hyperbilirubinemia. It becomes clinically apparent when serum bilirubin exceeds ~2-3 mg/dL.
Part 1: Bilirubin Metabolism
Step 1 - Bilirubin Production (Periphery)
Normal adults produce ~4 mg of bilirubin per kg body weight per day. 70-90% comes from hemoglobin degradation in senescent red blood cells, processed by the mononuclear phagocytic system (spleen, liver, bone marrow). The remainder comes from turnover of non-hemoglobin hemoproteins (myoglobin, cytochrome P-450, catalase, peroxidase).
Two-step conversion of heme to bilirubin:
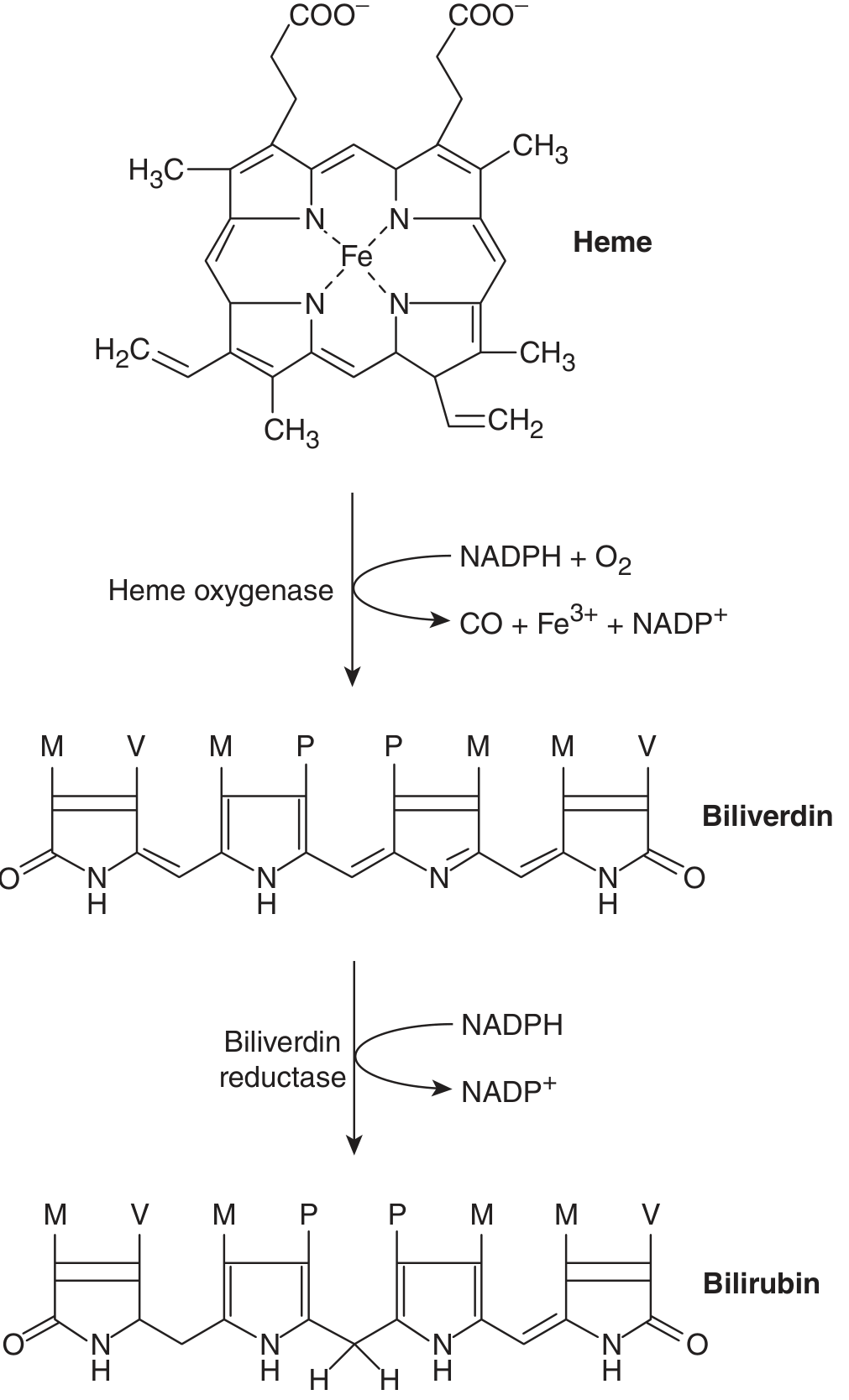
- Heme oxygenase cleaves the heme molecule at its alpha-bridge carbon → produces biliverdin (green tetrapyrrole) + CO + Fe³⁺. This reaction requires NADPH + O₂.
- Biliverdin reductase reduces biliverdin → bilirubin (yellow). This requires NADPH.
Note: Biliverdin cannot cross the placenta. Its reduction to bilirubin in mammals allows transplacental removal of fetal bilirubin into the maternal circulation. (Goldman-Cecil Medicine)
Step 2 - Transport to the Liver
Unconjugated bilirubin (UCB) is water-insoluble and is transported in plasma tightly bound to albumin. Certain drugs (salicylates, sulfonamides, furosemide, radiographic contrast agents) can competitively displace bilirubin from albumin - clinically important in neonates where free bilirubin increases the risk of kernicterus.
Step 3 - Hepatic Uptake and Conjugation
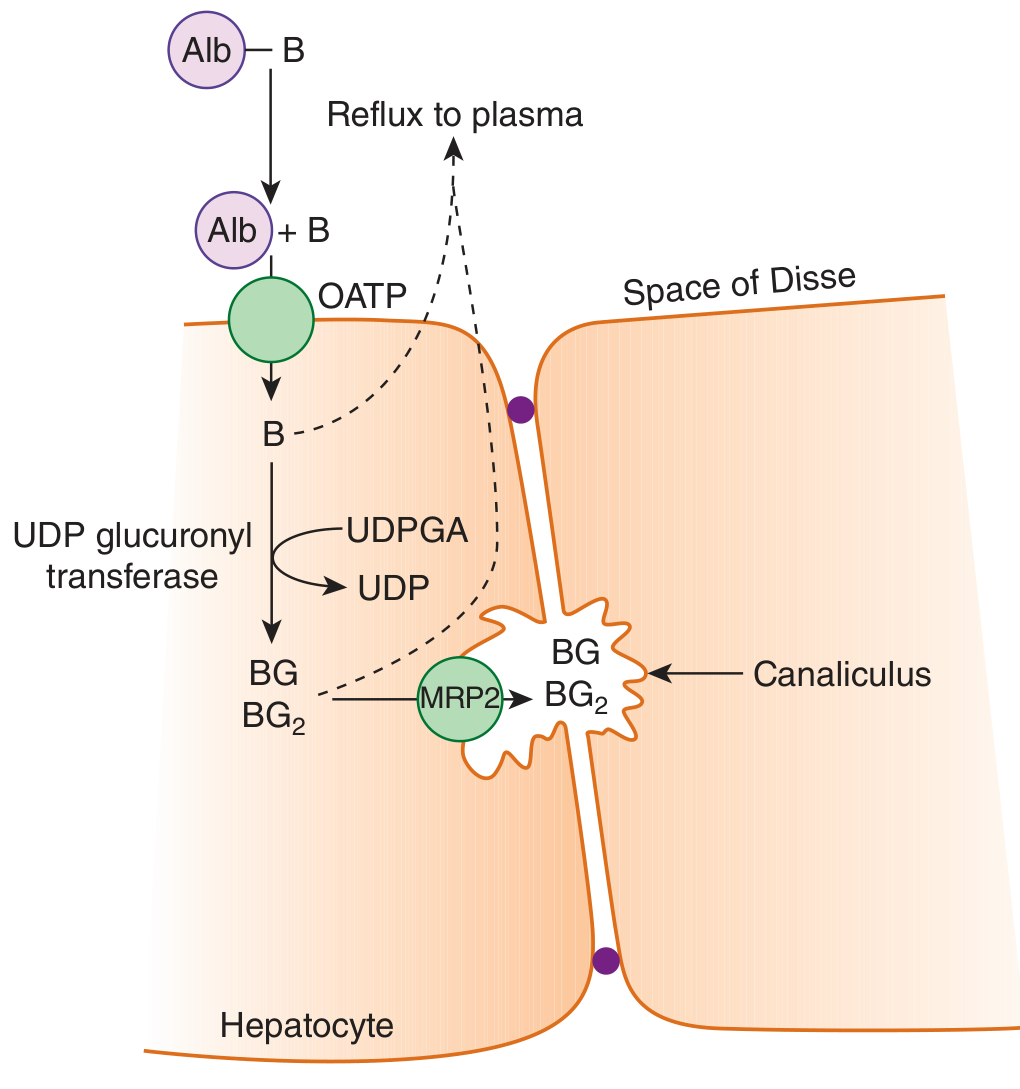
The liver processes bilirubin in four distinct steps:
| Step | Process | Key Players |
|---|---|---|
| 1 | Hepatocellular uptake | OATP (organic anion transporting polypeptide) family transporters; carrier-mediated, facilitated diffusion |
| 2 | Intracellular binding | Glutathione-S-transferases (ligandins) keep bilirubin in solution inside the hepatocyte |
| 3 | Conjugation | UDP-glucuronosyltransferase (UGT1A1) conjugates bilirubin with 1-2 glucuronic acid moieties → bilirubin monoglucuronide (BMG) and diglucuronide (BDG); makes it water-soluble |
| 4 | Biliary excretion | MRP2 (ABCC2) actively transports conjugated bilirubin across the canalicular membrane into bile; some refluxes back via MRP3 → OATP1B1/1B3 re-uptake (enterohepatic cycling) |
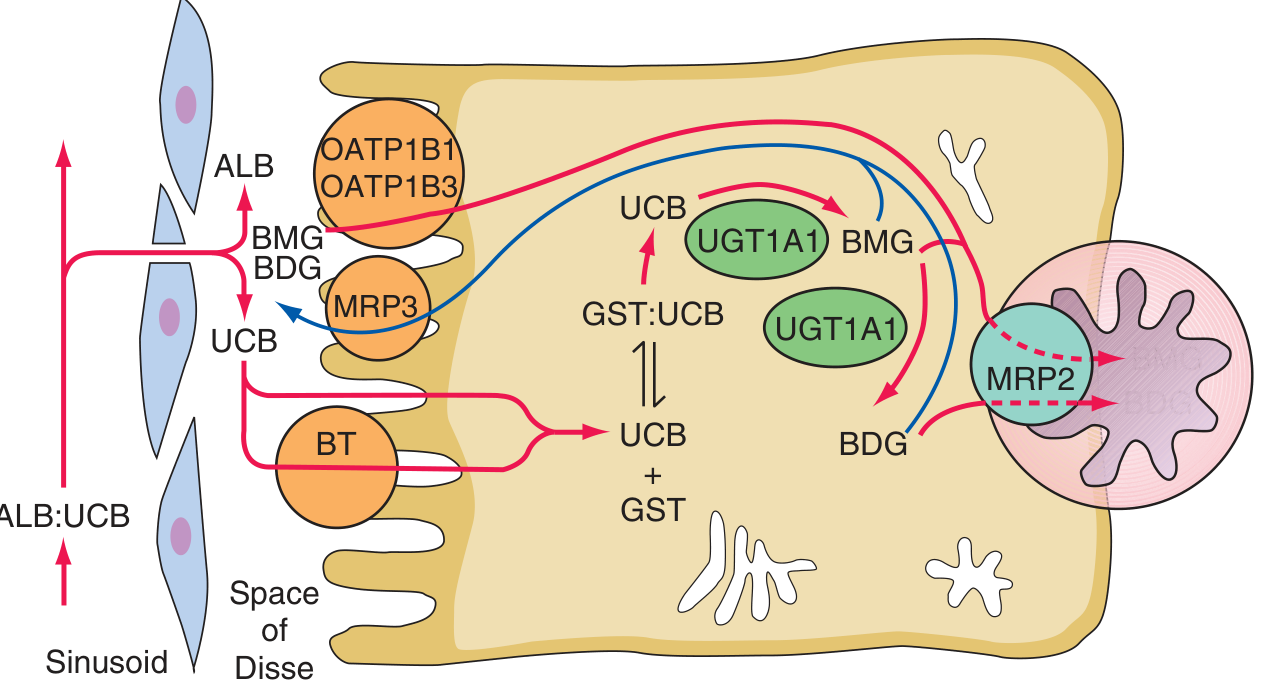
Step 4 - Intestinal Processing and Enterohepatic Circulation
- Conjugated bilirubin enters the intestine and cannot be reabsorbed (intestinal mucosa is relatively impermeable to it).
- Intestinal bacteria convert conjugated bilirubin → urobilinogen (colorless) → urobilin (brown, gives stool its color) and stercobilin.
- A portion of urobilinogen is reabsorbed into the portal circulation (enterohepatic circulation); some is re-excreted by the liver, small amounts enter systemic circulation and are excreted in urine (urobilinogen in urine = normal).
Step 5 - Renal Handling
- Unconjugated bilirubin: NOT excreted in urine (too tightly albumin-bound for glomerular filtration; no tubular secretion mechanism).
- Conjugated bilirubin: Readily filtered at the glomerulus → appears in urine ("bilirubinuria") in conditions with conjugated hyperbilirubinemia. Kidneys act as an "overflow valve" for conjugated bilirubin.
Part 2: Classification of Jaundice
Jaundice is classified by the site of the defect in bilirubin metabolism:
Type 1: Pre-Hepatic (Hemolytic) Jaundice
Mechanism: Excessive bilirubin production overwhelms normal hepatic processing.
| Feature | Detail |
|---|---|
| Bilirubin type | Predominantly unconjugated (indirect) |
| Urine bilirubin | Absent (unconjugated is albumin-bound, not filtered) |
| Urine urobilinogen | Markedly increased |
| Stool color | Dark (increased stercobilin) |
| Serum bilirubin | Rarely exceeds ~4 mg/dL in isolated hemolysis (liver can compensate up to ~8x normal RBC production); higher values suggest concurrent hepatic dysfunction |
| LFTs | Normal (AST, ALT, ALP normal) |
Causes:
- Hemolytic anemias (autoimmune, sickle cell, hereditary spherocytosis, G6PD deficiency)
- Malaria, transfusion reactions
- Ineffective erythropoiesis (thalassemia)
- Resorption of large hematomas
Type 2: Hepatic (Hepatocellular) Jaundice
Mechanism: Liver parenchymal damage impairs uptake, conjugation, and/or excretion of bilirubin.
| Feature | Detail |
|---|---|
| Bilirubin type | Both conjugated and unconjugated elevated |
| Urine bilirubin | Present (conjugated bilirubin spills into urine) |
| Urine urobilinogen | Variable (may be increased early, decreased in severe disease) |
| Stool color | Pale (reduced excretion) |
| LFTs | Elevated AST, ALT (hepatocyte damage); ALP mildly elevated |
Causes: Viral hepatitis (A, B, C), alcoholic hepatitis, cirrhosis, drug-induced hepatotoxicity, autoimmune hepatitis, liver failure.
Type 3: Post-Hepatic (Obstructive/Cholestatic) Jaundice
Mechanism: Obstruction of bile flow (intrahepatic or extrahepatic) prevents conjugated bilirubin from entering the gut.
| Feature | Detail |
|---|---|
| Bilirubin type | Predominantly conjugated (direct) |
| Urine bilirubin | Strongly positive ("dark urine") |
| Urine urobilinogen | Absent or markedly reduced (no bilirubin reaches gut) |
| Stool color | Pale/clay-colored (no stercobilin) |
| LFTs | Markedly elevated ALP and GGT; AST/ALT less prominent |
| Other | Pruritus (bile salt deposition in skin), steatorrhea, fat-soluble vitamin malabsorption |
Causes:
- Intrahepatic: Primary biliary cholangitis, primary sclerosing cholangitis, drugs, intrahepatic cholestasis of pregnancy
- Extrahepatic: Choledocholithiasis, pancreatic carcinoma, cholangiocarcinoma, biliary strictures, ampullary carcinoma
Part 3: Hereditary Disorders of Bilirubin Metabolism
These are isolated bilirubin disorders without other liver disease:
Unconjugated Hyperbilirubinemia
| Disorder | Defect | Bilirubin (mg/dL) | Phenobarbital response | Features |
|---|---|---|---|---|
| Crigler-Najjar Type I | Complete absence of UGT1A1 | 20-45 | None | AR; kernicterus; requires phototherapy 12 h/day; liver transplant is curative |
| Crigler-Najjar Type II | Severely reduced UGT1A1 (<10% normal) | 6-25 | Yes (significant reduction) | AR; generally survives without brain damage; responds to phenobarbital |
| Gilbert Syndrome | Mildly reduced UGT1A1 (10-33% normal; usually UGT1A1*28 promoter variant) | 1.5-4 | Yes | Very common (~5% population); benign; exacerbated by fasting, illness, stress |
Conjugated Hyperbilirubinemia
| Disorder | Gene/Protein | Mechanism | Key Feature |
|---|---|---|---|
| Dubin-Johnson Syndrome | ABCC2/MRP2 | Defective canalicular export of conjugated bilirubin | Black liver (lysosomal pigment); benign; coproporphyrin I in urine |
| Rotor Syndrome | SLCO1B1+SLCO1B3/OATP1B1+1B3 | Defective hepatic reuptake of conjugated bilirubin refluxed from MRP3 | No liver pigment; coproporphyrin I+III elevated |
| PFIC (Progressive Familial Intrahepatic Cholestasis) | ATP8B1 (FIC1), ABCB11 (BSEP), ABCB4 (MDR3) | Defective bile acid/lipid transport | Severe; progressive liver disease |
| BRIC (Benign Recurrent Intrahepatic Cholestasis) | ATP8B1 or ABCB11 | Same genes as PFIC, less severe mutations | Episodic, self-limited cholestasis |
Quick Diagnostic Summary
Jaundice
│
├── Unconjugated (indirect) dominant
│ ├── High urobilinogen, no bilirubinuria → PRE-HEPATIC (hemolysis)
│ └── Normal urobilinogen, no bilirubinuria → HEPATIC (conjugation defect: Gilbert, Crigler-Najjar)
│
└── Conjugated (direct) dominant
├── Both fractions elevated, bilirubinuria → HEPATOCELLULAR (hepatitis, cirrhosis)
└── Bilirubinuria + absent urobilinogen + pale stool → POST-HEPATIC (obstruction)
Sources:
- Harrison's Principles of Internal Medicine, 22nd Edition (2025), Ch. 349
- Goldman-Cecil Medicine, Ch. 133
- Ganong's Review of Medical Physiology, 26th Edition, Ch. 28
- Schwartz's Principles of Surgery, 11th Edition, Ch. 15
This is a shared conversation. Sign in to Orris to start your own chat.