helicobacter pylori: biochemical mechanism of mucosal injury
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Helicobacter pylori" AND "mucosal injury" AND mechanism
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive information from multiple authoritative sources. Let me compile the full answer.
Helicobacter pylori: Biochemical Mechanisms of Mucosal Injury
H. pylori causes mucosal injury through a coordinated set of mechanisms that can be grouped into: (1) colonization strategies, (2) direct toxin/enzyme effects, (3) immune-mediated injury, and (4) disruption of acid homeostasis.
1. Colonization and Initial Survival
Before any injury can occur, H. pylori must survive the hostile gastric environment. It does this via two critical adaptations:
Urease - Up to 15% of the organism's total protein is cytoplasmic urease. It converts periplasmic urea into CO₂ and ammonia (NH₃), which buffers the surrounding acid micro-environment. This local alkalinization lets the bacteria survive the gastric lumen long enough to burrow into the mucus layer. Strains lacking urease are nonpathogenic.
Flagella - Specialized flagella propel the organism from the gastric lumen, across the mucus gel, to the surface epithelium. Flagella-deficient strains are likewise nonpathogenic.
H. pylori resides predominantly in the deeper portions of the mucous gel or at the interface between the mucous layer and gastric epithelium. It typically does not invade the epithelial cell layer itself. - Harrison's Principles of Internal Medicine 22E, p. 2563
2. Adhesion and Direct Epithelial Injury
Once embedded near the epithelium, H. pylori attaches to gastric epithelial cells via specific adhesins:
- Neutrophil-activating protein A (NapA)
- Heat shock protein 60 (HspB)
- Sialic acid-binding adhesin (SabA)
- Outer membrane proteins (Hop proteins)
Attachment triggers direct injury and initiates the host inflammatory response. - Schwartz's Principles of Surgery 11E, p. 1145
3. Virulence Toxins - The Core Injury Mediators
Vacuolating Cytotoxin A (VacA)
VacA is secreted by most H. pylori strains and causes vacuolation within epithelial cells by forming anion-selective channels in the plasma membrane. This disrupts intracellular vesicular trafficking, induces mitochondrial apoptosis, and damages the epithelial barrier. The degree of vacuolation correlates with tissue damage. - Harrison's 22E
CagA (Cytotoxin-Associated Gene A) - the Major Oncogenic Virulence Factor
The cag pathogenicity island (cag-PAI) encodes a type IV secretion system that injects the CagA protein directly into host gastric epithelial cells. Once inside:
- CagA activates a cascade of cellular events controlling cell proliferation, cytokine production, and cytoskeletal rearrangement
- It disrupts normal cell polarity and tight junctions
- It promotes inflammatory signaling, contributing to gastritis and increasing carcinogenic risk
CagA-positive strains are significantly more associated with peptic ulceration and gastric cancer than CagA-negative strains. - Harrison's 22E, p. 2563; Bailey & Love 28E, p. 1176
4. Enzymatic Disruption of the Mucus Barrier
Beyond urease, H. pylori produces several enzymes that directly degrade mucosal defenses:
| Enzyme/Factor | Effect |
|---|---|
| Urease | Produces NH₃ - toxic to epithelium at high concentrations; disrupts mucus gel structure |
| Lipase | Degrades phospholipids in the mucus layer and cell membranes, reducing surface hydrophobicity |
| Catalase | Neutralizes host hydrogen peroxide (H₂O₂), helping evade oxidative killing |
| Platelet-activating factor (PAF) | Promotes local inflammation and vascular injury |
| PicB | Induces cytokine production in host cells |
The net result is disruption of the gastric mucous barrier, reducing the gel's protective properties and allowing luminal acid and pepsin to access the underlying epithelium. - Harrison's 22E; Bailey & Love 28E
5. Immune-Mediated Mucosal Injury
H. pylori does not simply destroy tissue by direct toxicity - the host immune response is a major driver of injury:
Acute phase: Recruitment of neutrophils is the initial inflammatory response. Neutrophils release reactive oxygen species (ROS) and reactive nitrogen species (RNS) that cause oxidative epithelial damage.
Chronic phase: Sequential accumulation of T and B lymphocytes, plasma cells, and macrophages follows. The resultant chronic gastric inflammation is characterized by:
- Enhanced mucosal expression of proinflammatory cytokines - IL-8, TNF-α, IL-1β
- Persistent generation of ROS and RNS
- Mucosal cell DNA damage and chromosomal instability
- Increased apoptosis
This immune-mediated injury progressively weakens mucosal defenses. Notably, H. pylori has evolved mechanisms to avoid complete destruction by the mucosal immune system, enabling it to persist for decades. - Schwartz's 11E, p. 1145
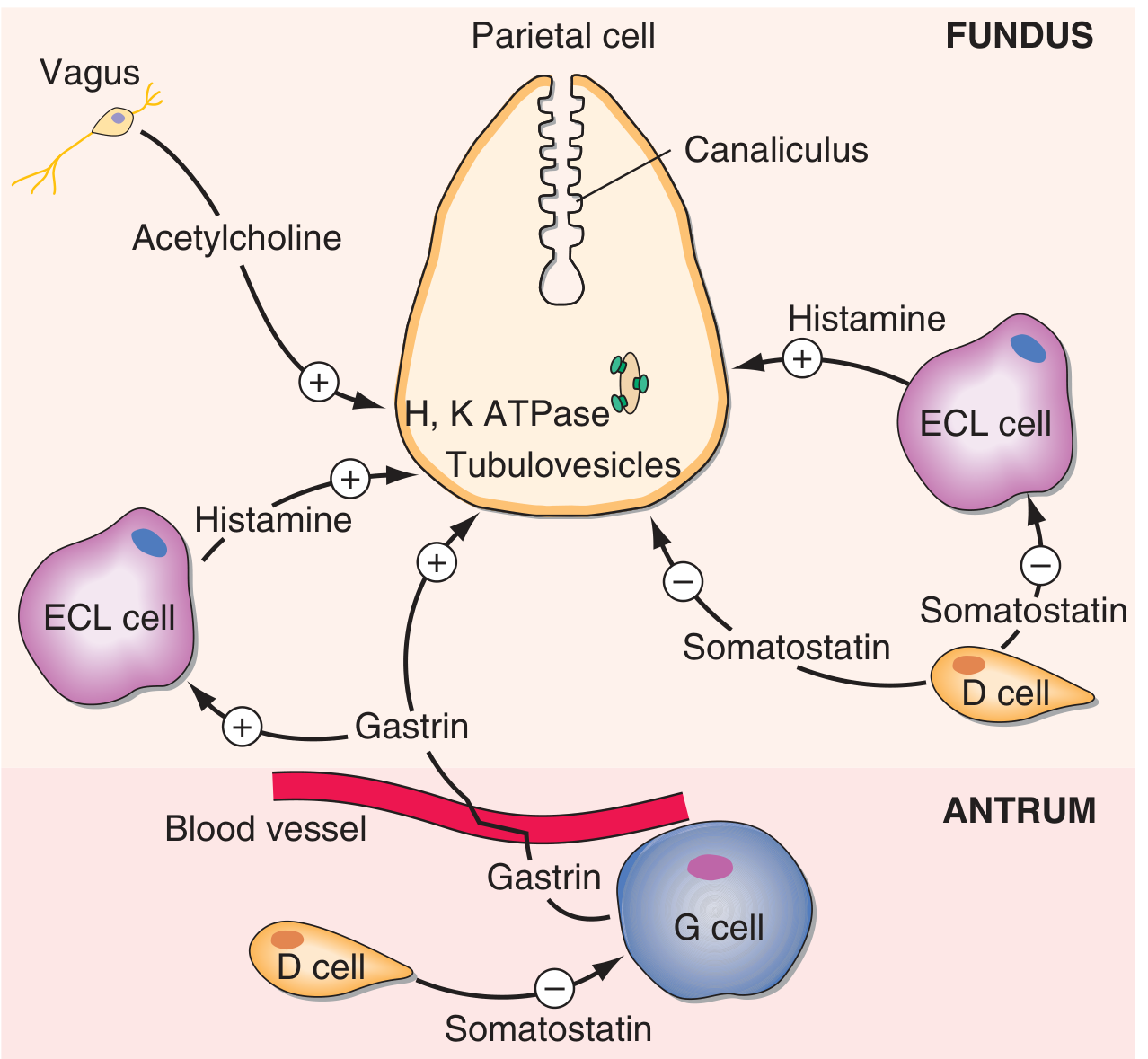
6. Gastric Acid Dysregulation - The Somatostatin-Gastrin Axis
This is the mechanism linking H. pylori to duodenal ulcer specifically:
Step-by-step sequence:
-
Ammonia from urease alkalinizes the antrum - normally, luminal acid stimulates D cells to release somatostatin, which inhibits gastrin from G cells. Ammonia blunts this feedback.
-
Depletion of antral somatostatin (D cells) - via three proposed mechanisms:
- Direct antral alkalinization removes the acid stimulus for somatostatin release
- Toxic cytokine effect on D cells
- H. pylori produces N-α-methylhistamine, an H₃ receptor agonist that binds H₃ receptors on D cells, directly suppressing somatostatin release
-
Unopposed G cell (gastrin) activity → hypergastrinaemia
-
Gastrin stimulates ECL cells → increased histamine release → parietal cell stimulation → acid hypersecretion and parietal cell hyperplasia
-
High duodenal acid load causes gastric metaplasia of the duodenum (a protective response)
-
Gastric metaplastic patches in the duodenum can now be colonized by H. pylori (normal duodenal mucosa cannot be infected). This produces local duodenitis and ultimately duodenal ulceration. The risk of duodenal ulcer increases 50-fold in patients with gastric metaplasia of the duodenum.
Additionally, H. pylori colonization of the duodenum causes a significant decrease in acid-stimulated duodenal bicarbonate production, removing a key mucosal defense against luminal acid.
In contrast, with long-standing infection involving the corpus (body), H. pylori can inhibit H⁺-K⁺-ATPase expression, leading to hypochlorhydria and atrophic gastritis - the pathway toward gastric cancer. - Schwartz's 11E, p. 1146; Harrison's 22E, p. 2564
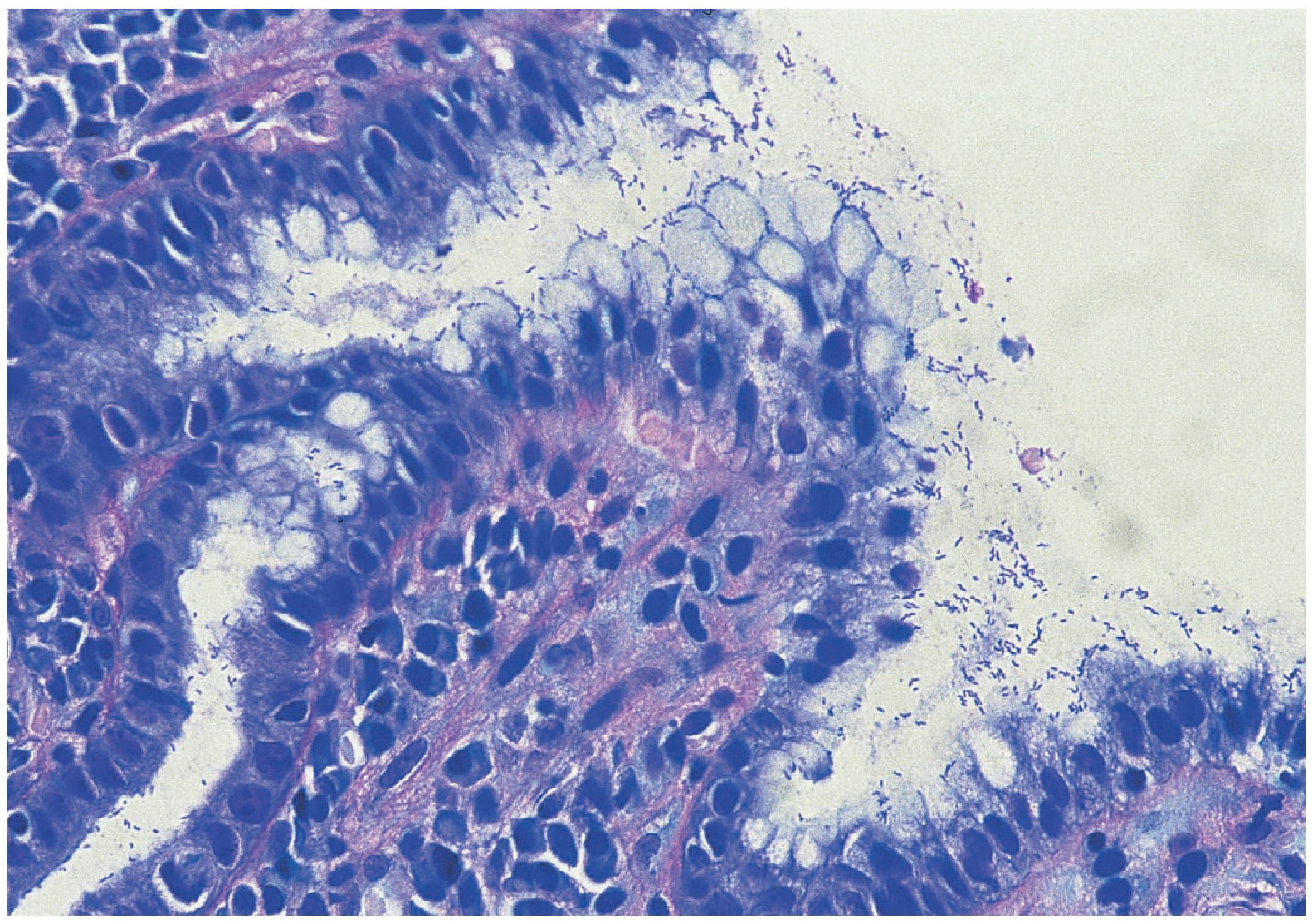
7. Histological Appearance of Mucosal Injury
Outcome Depends on Distribution of Gastritis
| Gastritis Pattern | Predominant Pathology |
|---|---|
| Antral-predominant (most common in the West) | Hypergastrinaemia, acid hypersecretion → duodenal ulcer |
| Corpus/pangastritis (more common in developing world) | Acid suppression, atrophy, intestinal metaplasia → gastric ulcer, gastric adenocarcinoma, MALT lymphoma |
Summary of Key Biochemical Events
H. pylori colonizes mucus layer
↓
Urease → NH₃ (local buffering + epithelial toxicity + antral alkalinization)
↓
Flagellar motility → epithelial adhesion (Hop proteins, NapA, HspB, SabA)
↓
Lipase + Urease → mucous barrier degradation
↓
VacA toxin → epithelial vacuolation → apoptosis
CagA (type IV SS) → intracellular signaling → cytokine production, DNA damage
↓
Neutrophil recruitment → ROS/RNS → oxidative epithelial damage
Macrophage/lymphocyte accumulation → IL-8, TNF-α, IL-1β → chronic inflammation
↓
Antral D cell suppression → ↓somatostatin → ↑gastrin → ↑acid
↓
Duodenal gastric metaplasia → H. pylori colonization
→ duodenal ulcer
Sources: Schwartz's Principles of Surgery 11E, pp. 1145-1146 | Harrison's Principles of Internal Medicine 22E, pp. 2563-2564 | Bailey & Love's Short Practice of Surgery 28E, pp. 1176-1177 | Medical Microbiology 9E
This is a shared conversation. Sign in to Orris to start your own chat.