Pheochromocytoma
Reading File
Finding Sources
Searching PubMed
pheochromocytoma diagnosis management
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive content from multiple authoritative textbooks, three pathology images, and recent PubMed evidence. I have enough to write a thorough, well-structured answer.
Pheochromocytoma
A pheochromocytoma is a catecholamine-secreting tumor arising from chromaffin cells of the adrenal medulla. Together with extra-adrenal paragangliomas, these tumors are collectively termed PPGLs (pheochromocytomas and paragangliomas). Though rare (estimated 2-8 cases per million per year), they are clinically important because they can cause fatal hypertensive crises, are potentially curable with surgery, and carry a significant hereditary component.
Epidemiology & the "Rule of 10s"
The traditional "rule of 10s" describes the distribution of pheochromocytomas:
- ~10% are extra-adrenal (paragangliomas)
- ~10% are bilateral
- ~10% occur in children
- ~10% are malignant at presentation (more recent data suggests this may exceed 10%)
- ~10% are familial (this figure has substantially increased - now estimated >30-40% carry germline mutations)
Pheochromocytomas are found in 0.2%-0.6% of hypertensive patients, and may be more common in hypertensive children (~1.7%). They are discovered incidentally or, in tragic cases, only at autopsy. - Brenner and Rector's The Kidney
Pathology
Gross Morphology
Pheochromocytomas range from small circumscribed lesions to large hemorrhagic masses; the average weight is ~100 g (range: >1 g to nearly 4,000 g). Smaller tumors are yellow-tan on cut section; larger ones tend to be hemorrhagic, necrotic, and cystic. A key histochemical feature: incubation with potassium dichromate turns the tumor dark brown due to oxidation of stored catecholamines - the origin of the term "chromaffin."
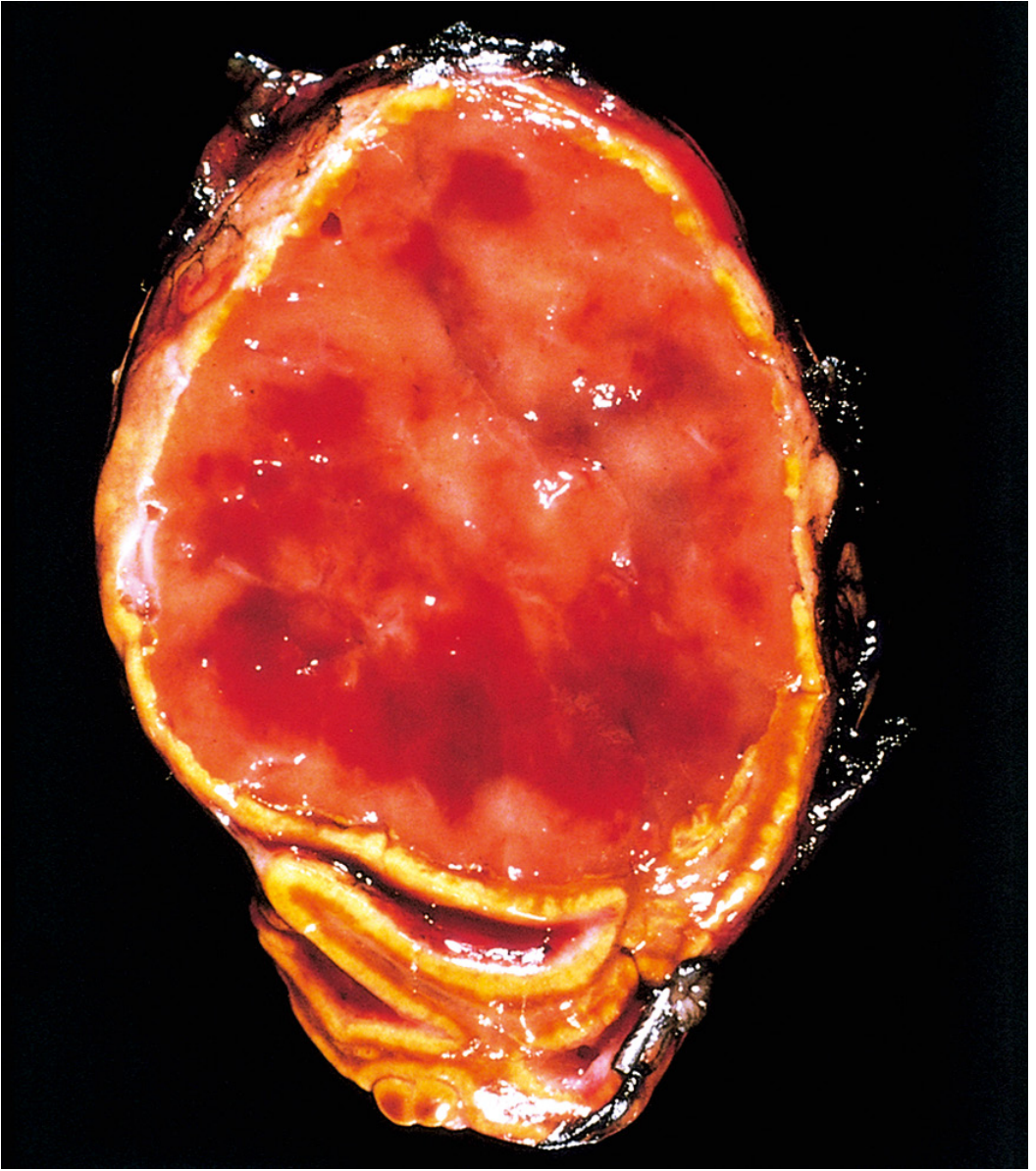
Histology
The classic pattern is the "zellballen" (German: "cell balls") - clusters of polygonal to spindle-shaped chromaffin cells (chief cells) surrounded by sustentacular cells, arranged in small nests or alveoli supplied by a rich vascular network. The cytoplasm has a finely granular appearance due to catecholamine-containing granules. Nuclei show characteristic "salt-and-pepper" chromatin typical of neuroendocrine tumors. Electron microscopy reveals membrane-bound, electron-dense secretory granules.
Immunohistochemistry:
- Chief cells: positive for chromogranin and synaptophysin
- Sustentacular cells: positive for S-100
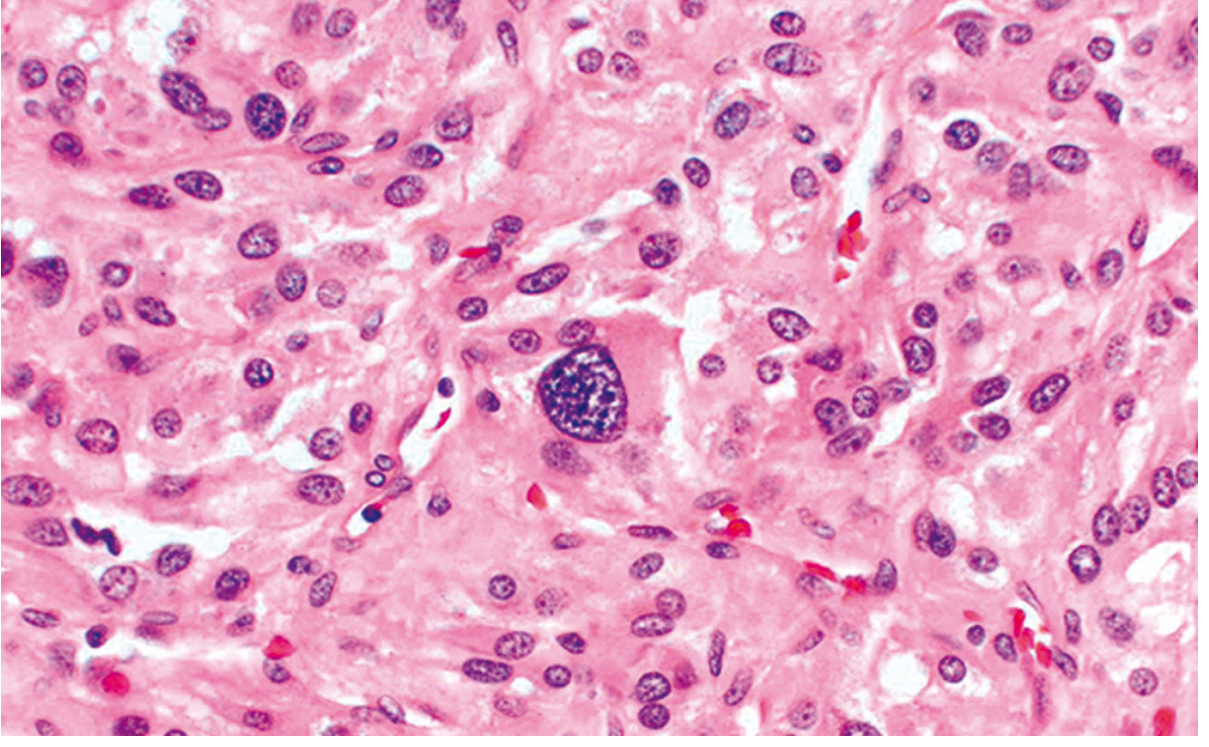
Determining Malignancy
Malignancy cannot be determined histologically. Cellular pleomorphism, mitoses, and even vascular/capsular invasion can be seen in benign tumors. Paradoxically, cellular monotony may be associated with aggressive behavior. The only reliable indicator of malignancy is the presence of metastases - to regional lymph nodes, liver, lung, or bone. - Robbins, Cotran & Kumar Pathologic Basis of Disease
Hereditary Syndromes & Genetics
More than 30% of pheochromocytomas are now associated with germline mutations. Key syndromes include:
| Syndrome | Gene | Key Features |
|---|---|---|
| MEN2A | RET proto-oncogene | Pheo (~50%, usually bilateral), medullary thyroid carcinoma, parathyroid adenomas, cutaneous lichen amyloidosis |
| MEN2B | RET | Bilateral pheo, medullary thyroid Ca, submucosal neuromas, marfanoid body habitus, Hirschsprung disease |
| Von Hippel-Lindau (VHL) type 2 | VHL (chr 3p25-26) | Pheo/paraganglioma in ~20%, retinal/cerebellar hemangioblastomas, clear cell RCC, pancreatic NET |
| Neurofibromatosis type 1 | NF1 (chr 17q11.2) | Pheo in ~2%, neurofibromas, café-au-lait spots, Lisch nodules |
| Familial paraganglioma | SDHB (chr 1p35-36), SDHD (chr 11q23) | Paragangliomas of skull base, neck, thorax, abdomen, bladder |
| Sturge-Weber | GNAQ | Port wine stain, leptomeningeal angiomas |
Current guidelines recommend genetic testing for all patients with pheochromocytoma, with a shared decision-making process often involving family members. - Brenner and Rector's The Kidney
Molecular Clusters
Tumors are broadly divided into two molecular clusters based on secretory behavior:
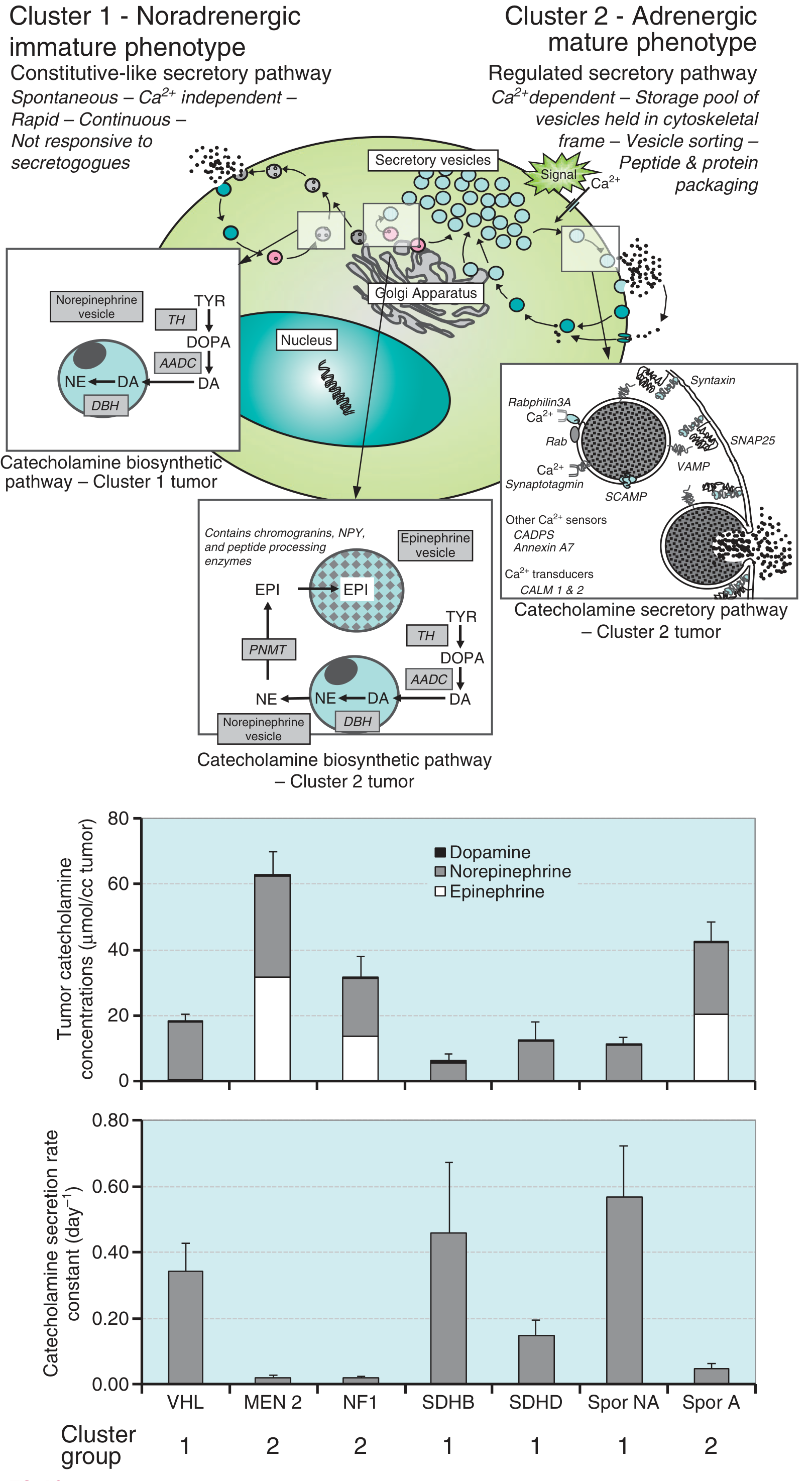
- Cluster 1 (noradrenergic): VHL, SDHB, SDHD mutations - constitutive, Ca2+-independent secretory pathway; predominantly norepinephrine
- Cluster 2 (adrenergic): MEN2, NF1 - regulated, Ca2+-dependent secretory pathway; produce both epinephrine and norepinephrine
Clinical Features
Symptoms
The classic triad is: headache, sweating, and hypertension (reported in ~95% in large series). Other features include:
- Tachycardia and palpitations
- Tremor and sense of apprehension
- Abdominal or chest pain, nausea, vomiting
- Pallor (due to vasoconstriction)
Hypertension is present in ~90% of patients. Approximately two-thirds experience paroxysmal episodes - abrupt, precipitous BP elevation superimposed on chronic hypertension. Isolated paroxysms without sustained hypertension occur in less than half.
Precipitants of paroxysms: emotional stress, exercise, posture change, tumor palpation, micturition (bladder paragangliomas), certain drugs (metoclopramide, beta-blockers, glucocorticoids, histamine, tyramine).
Cardiac Complications
- Congestive heart failure, pulmonary edema, myocardial infarction, ventricular fibrillation, stroke
- Catecholamine cardiomyopathy: focal myocardial necrosis, mononuclear infiltrates, and interstitial fibrosis due to catecholamine-induced vasospasm or direct catecholamine toxicity
- One of the most dangerous presentations is unexplained shock, possibly followed by multisystem organ failure
- Pheo in pregnancy masquerading as preeclampsia carries high maternal and fetal mortality if undiagnosed
Differential Diagnosis
Many conditions mimic pheochromocytoma:
- Anxiety disorders and panic attacks
- Essential hypertension
- Hyperthyroidism
- Carcinoid syndrome
- Systemic mastocytosis
- Migraine/cluster headaches
- Baroreflex failure, POTS
- Factitious (self-administered catecholamines)
Clues from physical exam: café-au-lait spots/neurofibromas (NF1), retinal hemangiomas (VHL), port wine stain (Sturge-Weber), subungual fibromas/ash leaf patches (tuberous sclerosis), marfanoid habitus (MEN2B). - Brenner and Rector's The Kidney
Biochemical Diagnosis
The first critical step is simply thinking of the tumor. Given the rarity and many mimics, clinical suspicion must be high.
Preferred tests:
- Plasma free metanephrines - highest sensitivity; brief snapshot of catecholamine production and metabolism. Preferred when rapid, accurate assay is available.
- 24-hour urinary fractionated metanephrines - integrates catecholamine production over time; more widely available and less costly.
Why metanephrines rather than catecholamines themselves?
Pheochromocytomas continuously O-methylate catecholamines to metanephrines within the tumor (even between episodic secretory bursts), so metanephrines provide more consistent elevation than the parent catecholamines.
Causes of false-negative results: familial tumors, normotensive tumors, dopamine-beta-hydroxylase deficiency, intermittently secreting tumors.
Pharmacologic testing (used in equivocal cases):
- Clonidine suppression test (preferred) - failure of clonidine to suppress plasma norepinephrine supports the diagnosis
- Glucagon stimulation (rarely used due to risk)
The definitive diagnosis of malignancy requires evidence of metastasis. - Tietz Textbook of Laboratory Medicine, 7th Edition
Imaging & Localization
Imaging is ordered ONLY after biochemical confirmation - to improve cost-effectiveness and reduce radiation exposure.
| Modality | Notes |
|---|---|
| CT (adrenal, thin cuts) | First-line; high resolution for adrenal and peri-adrenal tissue |
| MRI | Preferred for extra-adrenal, pelvic, bladder locations; avoids radiation; pheo appears bright on T2 |
| 123I-MIBG scintigraphy | Functional imaging; useful for locating extra-adrenal and metastatic disease; confirms catecholamine-synthesizing tissue |
| FDG-PET | Particularly useful for SDHB-mutated tumors and metastatic disease |
| 68Ga-DOTATATE PET | Increasingly used for paragangliomas |
Extra-adrenal paragangliomas most commonly occur at the organ of Zuckerkandl (aortic bifurcation) or near the bladder. - Brenner and Rector's The Kidney
Treatment
Preoperative Preparation (Critical)
Proper pharmacologic preparation is essential for safe surgery:
- Alpha-blockade first - phenoxybenzamine (long-acting, irreversible) orally, or phentolamine IV. Start 7-14 days before surgery to normalize BP, HR, and intravascular volume.
- Beta-blockers second (only after adequate alpha-blockade is established) - to control tachycardia. Never start beta-blockers alone, as this can cause paradoxical hypertension from unopposed alpha-stimulation.
- Volume expansion - liberal salt and fluid intake pre-operatively to prevent postoperative hypotension.
- Calcium channel blockers - often added before beta-blockers.
- Metyrosine (tyrosine hydroxylase inhibitor) - occasionally used for very large tumors or nonsurgical candidates to reduce catecholamine synthesis.
Intraoperative Management
- Anticipate hypertensive episodes during tumor manipulation
- IV agents with rapid onset/short half-life: nitroprusside, phentolamine, nitroglycerin, nicardipine
- Short-acting beta-blockers: labetalol, esmolol
- Aggressive fluid management upon tumor removal to counter sudden loss of tonic vasoconstriction
Postoperative Management
- Monitor for hypotension (guided by invasive monitoring; phenylephrine and fluids)
- Correct hypoglycemia (rebound from loss of catecholamine-mediated glycogenolysis)
- Electrolyte correction
- Persistent hypertension is not uncommon and may require continued antihypertensives
Surgical Approach
- Laparoscopic adrenalectomy is preferred for small, localized adrenal pheochromocytomas
- Open approach for large or locally invasive tumors
- Recent evidence: A 2025 systematic review and meta-analysis (PMID 40214691) examined outcomes of partial adrenalectomy for pheochromocytoma, relevant for bilateral or hereditary cases to preserve adrenocortical function.
Follow-Up
- Repeat biochemical testing 4-6 weeks post-operatively to confirm complete resection
- Annual or individualized long-term surveillance, especially with hereditary syndromes
- Metastatic disease: treated with MIBG therapy, sunitinib, chemotherapy (cyclophosphamide/vincristine/dacarbazine regimen), or PRRT (peptide receptor radionuclide therapy) - Campbell Walsh Wein Urology; Brenner and Rector's The Kidney
Summary Table
| Feature | Detail |
|---|---|
| Origin | Adrenal medulla chromaffin cells |
| Incidence | 2-8/million/year |
| Classic triad | Headache, sweating, hypertension |
| Hypertension | ~90% of patients |
| Malignancy diagnosis | Metastases only (histology unreliable) |
| Preferred biochemical test | Plasma free metanephrines or 24-hr urinary fractionated metanephrines |
| First-line imaging | CT (after biochemical confirmation) |
| Key preop drug | Alpha-blocker (phenoxybenzamine) FIRST |
| Surgery | Laparoscopic adrenalectomy (preferred for small tumors) |
| Hereditary proportion | >30% carry germline mutations |
| Malignancy marker | Distant metastases (lymph nodes, liver, lung, bone) |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Brenner and Rector's The Kidney; Tietz Textbook of Laboratory Medicine, 7th Edition; Campbell Walsh Wein Urology; Fuster and Hurst's The Heart, 15th Edition.
This is a shared conversation. Sign in to Orris to start your own chat.