How dna repair occurs in detail
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Reading File
Reading File
Reading File
I now have all the content needed for a comprehensive, well-illustrated answer. Let me compile the complete response.
DNA Repair: A Detailed Overview
DNA in every cell is under constant assault - from normal metabolic byproducts (reactive oxygen species, spontaneous deamination) to exogenous agents (UV radiation, ionizing radiation, chemical carcinogens). An estimated thousands of lesions per cell per day must be corrected to maintain genomic integrity. Without repair, permanent mutations accumulate, driving cancer and inherited disease. Cells employ multiple, overlapping repair systems, each specialized for a particular type of damage.
Why DNA Repair Is Needed: Sources of Damage
| Damaging Agent | Type of Damage |
|---|---|
| UV radiation (sunlight) | Pyrimidine (thymine) dimers, cyclobutane photoproducts |
| Ionizing radiation (X-rays) | Double-strand breaks, hydroxyl radical damage |
| Chemical carcinogens (e.g., benzo[a]pyrene from smoke) | Bulky G adducts, helix distortion |
| Reactive oxygen species | Base oxidation, strand breaks |
| Alkylating agents (e.g., nitrous acid, dimethyl sulfate) | O6-methylguanine, base deamination |
| Spontaneous hydrolysis | Depurination (~10,000 purine losses/cell/day), cytosine deamination to uracil |
| Replication errors | Mismatched base pairs |
"DNA is constantly being subjected to environmental insults that cause the alteration or removal of nucleotide bases... if the damage is not repaired, a permanent change (mutation) is introduced that can result in any of a number of deleterious effects, including loss of control over the proliferation of the mutated cell, leading to cancer." - Biochemistry, 8th ed., Lippincott Illustrated Reviews
Overview: General Repair Steps
Most excision-based repair systems share the same core logic:
- Recognition - A sensor protein detects the lesion (distorted helix, mismatched base, oxidized base)
- Excision - An endonuclease nicks the strand; an exonuclease removes the damaged fragment
- Gap filling - DNA polymerase synthesizes new DNA using the intact complementary strand as template (5'→3' direction)
- Ligation - DNA ligase seals the remaining nick
The exception is direct repair, where the damage is chemically reversed without removing any nucleotides.
Repair Mechanism 1: Direct Repair
The simplest form - repairs a lesion in one step without strand excision.
Example: O6-Methylguanine DNA Methyltransferase (MGMT)
- Alkylating agents add methyl or ethyl groups to the O6 position of guanine, causing it to mispair with thymine instead of cytosine
- MGMT transfers the alkyl group from the damaged base directly onto a cysteine in its own active site, restoring normal guanine
- The enzyme is consumed ("suicide enzyme") - one molecule per repair event
Example: Photoreactivation (bacteria and lower eukaryotes)
- Photolyase enzyme binds the pyrimidine dimer and uses energy from visible light (300-500 nm) to cleave the covalent bonds between the fused pyrimidines
- Restores the individual bases without removing any nucleotides
- Not present in humans
Repair Mechanism 2: Mismatch Repair (MMR)
When: Immediately after DNA replication - catches errors that escape polymerase proofreading
What it fixes: Mispaired bases (e.g., G-T), small insertions/deletions in microsatellite sequences
Steps in E. coli (and analogous process in humans):
Step 1 - Strand discrimination:
The repair machinery must identify which strand is the "new" (incorrect) strand and which is the parental template. In prokaryotes, this is based on methylation:
- GATC sequences on the parental strand are methylated on adenine by DNA adenine methylase (DAM)
- Immediately after replication, the daughter strand is unmethylated (hemimethylated state)
- MutS protein recognizes the mismatch; MutL is recruited; together they activate MutH, which specifically cleaves the unmethylated (daughter) strand
Step 2 - Excision:
An exonuclease removes nucleotides from the nicked daughter strand, including and beyond the mismatched base, leaving a gap
Step 3 - Resynthesis and ligation:
DNA pol III fills the gap using the parental strand as template; DNA ligase seals the nick
In humans: MutS homologues (MSH2, MSH6) and MutL homologues (MLH1, PMS2) perform the same function. The mechanism for identifying the new strand likely involves nicks present in newly synthesized DNA.
MMR reduces replication error rate from 1 in 10^7 to 1 in 10^9 nucleotides.
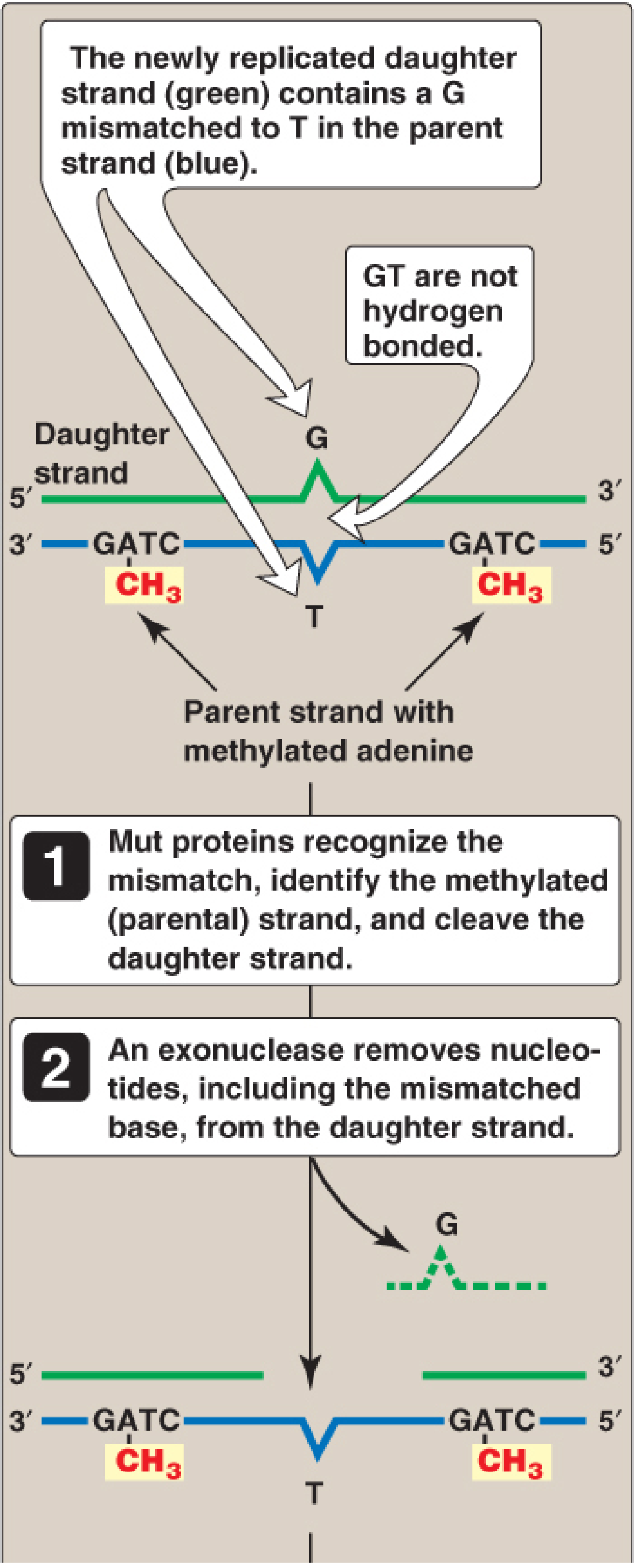
Clinical relevance: Mutations in MMR genes (especially MSH2 and MLH1) cause Lynch syndrome (HNPCC - Hereditary Non-Polyposis Colorectal Cancer), accounting for ~5% of all colon cancer. These tumors show microsatellite instability (MSI).
Repair Mechanism 3: Nucleotide Excision Repair (NER)
When: Any time; occurs throughout the cell cycle
What it fixes: Bulky, helix-distorting lesions - thymine dimers (UV-induced), large chemical adducts (benzo[a]pyrene-G adducts), intrastrand cross-links
NER removes a fragment of ~24-34 nucleotides containing the lesion.
Two NER sub-pathways:
| Sub-pathway | Trigger | Where |
|---|---|---|
| Global Genome Repair (GGR) | XPC-RAD23B complex detects helix distortion | Anywhere in genome |
| Transcription-Coupled Repair (TCR) | RNA polymerase stalls at a lesion | Actively transcribed genes |
Transcribed genes are repaired faster than the rest of the genome via TCR - this ensures that actively expressed genes are prioritized.
Steps in detail (human NER):
- Damage recognition
- GGR: XPC-RAD23B (with XPE/DDB2 complex) scans the genome and detects helix distortion
- TCR: RNA polymerase stalls at the lesion; CSA and CSB proteins are recruited to signal repair
- Helix unwinding
- Transcription factor TFIIH is recruited; its helicase subunits XPB (3'→5') and XPD (5'→3') unwind ~25-30 base pairs around the lesion
- XPA stabilizes the open DNA complex and helps verify the damage
- Dual incision
- XPF-ERCC1 nuclease cuts the damaged strand on the 5' side of the lesion
- XPG nuclease cuts the damaged strand on the 3' side
- A 24-34 nucleotide oligonucleotide containing the lesion is released
- Gap filling and ligation
- PCNA (sliding clamp), RPA (single-strand binding protein), and DNA polymerase δ/ε fill the gap using the intact complementary strand as template
- DNA ligase seals the final nick
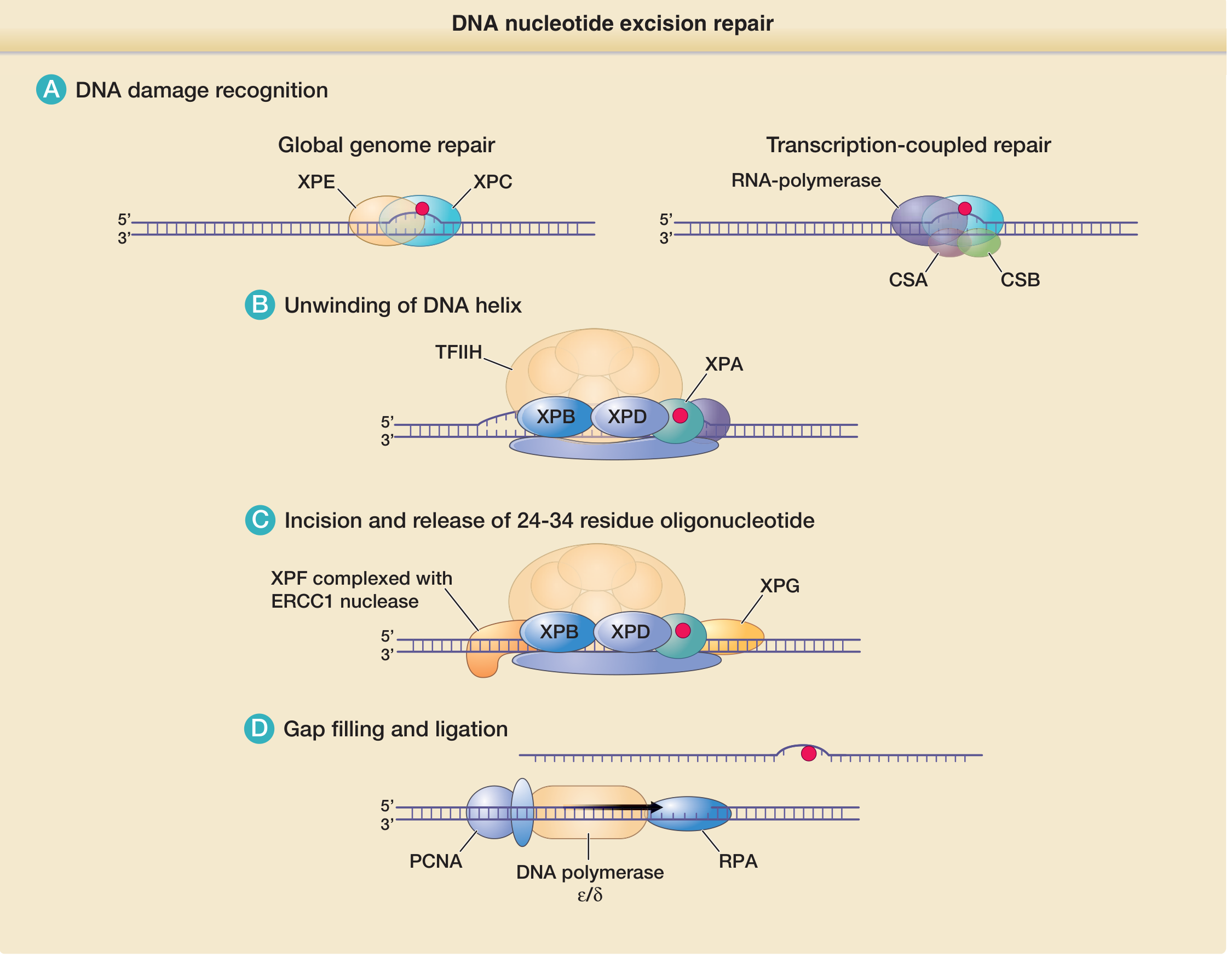
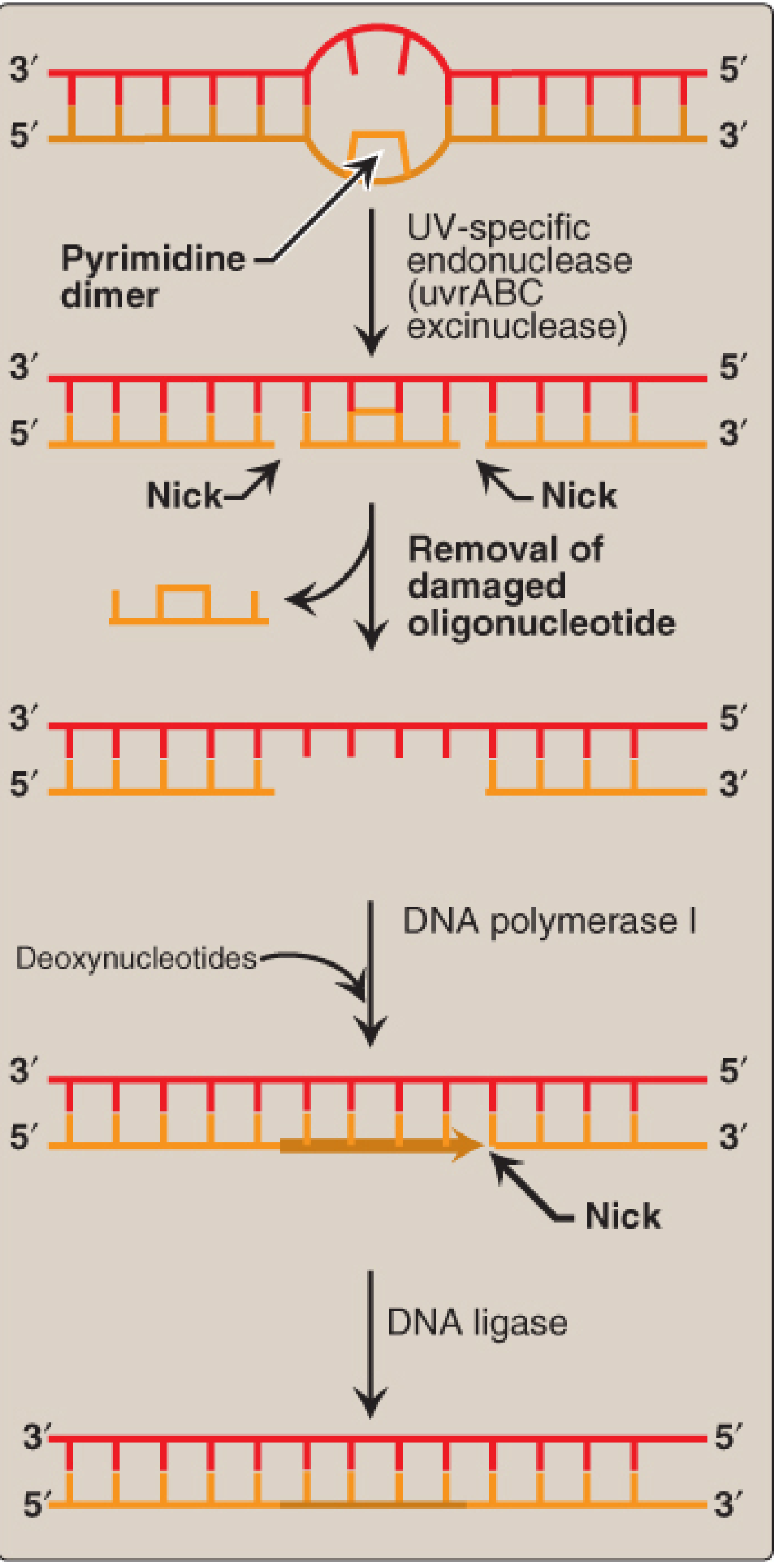
Clinical relevance:
- Xeroderma Pigmentosum (XP): Autosomal recessive; caused by mutations in any of 7 XP genes (XP-A through XP-G). Patients have extreme UV sensitivity and develop skin cancers very early in life.
- Cockayne Syndrome (CS): Mutations in CSA or CSB, affecting transcription-coupled repair specifically. Characterized by premature aging, neurodegeneration, and UV sensitivity without skin cancer.
- Trichothiodystrophy (TTD): Mutations in XPD/XPB; hair, nail, and skin abnormalities.
Repair Mechanism 4: Base Excision Repair (BER)
When: Any time
What it fixes: Small, non-helix-distorting lesions - oxidized bases, alkylated bases, spontaneous deamination products (e.g., uracil in DNA), abasic (AP) sites, single-strand breaks
BER is the most frequently used repair pathway.
Steps in detail:
Step 1 - Damaged base recognition and removal:
- Specific DNA glycosylases recognize the abnormal base and cleave the N-glycosidic bond between the base and the deoxyribose sugar
- This releases the abnormal base and leaves an abasic (AP) site - an apurinic or apyrimidinic site
- Examples: Uracil-DNA glycosylase removes uracil (from C deamination); OGG1 removes 8-oxoguanine (oxidative damage); MYH glycosylase removes adenine misincorporated opposite 8-oxoG
Step 2 - AP site processing:
- AP endonuclease (APE1) cleaves the phosphodiester backbone 5' to the AP site, creating a nick with a 3'-OH and a 5'-deoxyribose phosphate (dRP)
- DNA polymerase β (in short-patch BER) removes the dRP group using its lyase activity and inserts one correct nucleotide
Step 3 - Gap filling and ligation:
- Short-patch BER (most common): Pol β inserts a single nucleotide; XRCC1-DNA ligase III seals the nick
- Long-patch BER: Pol δ/ε displaces 2-10 nucleotides (strand displacement synthesis); the displaced "flap" is cleaved by FEN1; DNA ligase I seals the nick
Clinical relevance:
- Mutations in MYH (the DNA glycosylase that removes adenine mispaired with 8-oxoguanine) cause MAP - MUTYH-associated polyposis, an autosomal recessive form of multiple colorectal adenomas/cancer
Repair Mechanism 5: Double-Strand Break (DSB) Repair
When: In response to ionizing radiation, chemotherapy (topoisomerase poisons), and certain normal physiological processes (V(D)J recombination, meiosis)
What it fixes: Complete breaks through both strands of the DNA duplex - the most dangerous type of damage
There are two major pathways:
5a. Homologous Recombination (HR)
- Requires a homologous DNA template (sister chromatid or homologous chromosome)
- Therefore only active in S and G2 phases of the cell cycle (after replication)
- High fidelity - restores the exact sequence
Steps:
- End resection: The MRN complex (MRE11-RAD50-NBS1) and CtIP resect the broken ends to produce long 3' single-stranded overhangs
- RPA coating: RPA (replication protein A) coats the single-stranded DNA to protect it
- RAD51 loading: BRCA2 facilitates loading of RAD51 recombinase onto the ssDNA, displacing RPA. BRCA1 helps recruit the repair machinery.
- Strand invasion: The RAD51-coated single strand invades the homologous duplex (usually the sister chromatid), forming a D-loop (displacement loop)
- DNA synthesis: DNA polymerase extends from the 3'-OH end using the sister chromatid as template
- Resolution: The Holliday junction intermediates are resolved by resolvases/dissolverases, restoring two intact duplexes
Clinical relevance: Mutations in BRCA1 or BRCA2 impair HR. This dramatically increases the risk of hereditary breast and ovarian cancer. This also creates a vulnerability - cells with BRCA1/2 mutations become dependent on PARP for survival, which is the basis of PARP inhibitor therapy (olaparib, rucaparib).
5b. Non-Homologous End Joining (NHEJ)
- Does not require a homologous template
- Active in all phases of the cell cycle (especially G1)
- Error-prone - can cause small insertions or deletions (indels) at the junction
- The predominant DSB repair pathway in mammals
Steps:
- End recognition: Ku70/Ku80 heterodimer rapidly binds both broken DNA ends and protects them from degradation
- Recruitment of DNA-PKcs: The Ku complex recruits DNA-dependent protein kinase catalytic subunit (DNA-PKcs), forming the DNA-PK holoenzyme. DNA-PK synapses the two ends together.
- End processing: Nucleases (Artemis, activated by DNA-PKcs) and polymerases (pol μ, pol λ) trim and fill in the ends as needed to make them compatible for joining
- Ligation: The XRCC4-DNA Ligase IV complex (with XLF/Cernunnos) ligates the two ends together
Normal physiology: NHEJ is essential for V(D)J recombination during lymphocyte development (immune diversity). Defects in NHEJ components cause severe combined immunodeficiency (SCID) and radiosensitivity.
Repair Mechanism 6: Translesion Synthesis (TLS) - Damage Tolerance
When damage cannot be repaired before replication begins, specialized translesion synthesis (TLS) polymerases can bypass the lesion rather than repair it:
- Normal replicative polymerases stall at bulky lesions
- TLS polymerases (pol η, pol ι, pol κ, REV1) have larger, more flexible active sites that can accommodate distorted templates
- They insert nucleotides across the lesion, often with reduced fidelity (mutagenic bypass)
- Pol η specifically inserts AA opposite thymine dimers - this is essentially error-free bypass
- Xeroderma Pigmentosum Variant (XP-V) is caused by mutations in pol η - patients can repair dimers via NER but cannot perform error-free bypass, leading to increased UV mutagenesis
Integration with the Cell Cycle: The DNA Damage Response (DDR)
DNA repair does not occur in isolation - it is tightly coordinated with cell cycle progression:
- DNA damage is sensed by the MRN complex (DSBs) or RPA-loaded ssDNA (replication stress)
- ATM kinase (activated by DSBs) or ATR kinase (activated by ssDNA/stalled replication forks) are recruited
- ATM/ATR phosphorylate and activate checkpoint kinases CHK2 and CHK1, respectively
- These kinases phosphorylate CDC25 phosphatases, inactivating them, which prevents activation of cyclin-CDK complexes - cell cycle arrest
- ATM/ATR also activate p53 (TP53), which transcriptionally induces p21 (a CDK inhibitor) for sustained arrest, and BAX/PUMA for apoptosis if damage is irreparable
- Repair then proceeds; once complete, the checkpoint is lifted and the cell cycle resumes
Summary Table
| Pathway | Lesion Repaired | Key Proteins | Disease if Defective |
|---|---|---|---|
| Direct repair | O6-alkylguanine, cyclobutane dimers (bacteria) | MGMT, photolyase | - |
| Mismatch Repair (MMR) | Replication mismatches, small indels | MSH2, MSH6, MLH1, PMS2 | Lynch syndrome (HNPCC) |
| Nucleotide Excision Repair (NER) | Thymine dimers, bulky adducts | XPA-XPG, TFIIH, ERCC1 | Xeroderma pigmentosum, Cockayne syndrome, TTD |
| Base Excision Repair (BER) | Oxidized/alkylated/deaminated bases, AP sites | DNA glycosylases, APE1, Pol β, XRCC1 | MUTYH-associated polyposis |
| Homologous Recombination (HR) | Double-strand breaks (S/G2) | BRCA1/2, RAD51, MRN complex | Hereditary breast/ovarian cancer |
| Non-Homologous End Joining (NHEJ) | Double-strand breaks (all phases) | Ku70/80, DNA-PKcs, Ligase IV | SCID, radiosensitivity |
| Translesion Synthesis (TLS) | Bypass of unrepaired lesions | Pol η, pol ι, pol κ, REV1 | XP Variant (XP-V) |
Sources:
- Biochemistry, 8th ed., Lippincott Illustrated Reviews, Ch. 30 (DNA repair, pp. 1176-1187)
- Basic Medical Biochemistry: A Clinical Approach, 6e, Ch. 12 (DNA damage & repair, pp. 411-417)
- Emery's Elements of Medical Genetics and Genomics, p. 34 (Table 2.7 - DNA repair pathways)
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Ch. 68 (Table 68.3 - DNA Repair Pathways)
- Campbell-Walsh-Wein Urology, Ch. 33 (DNA Damage Response)
- Fitzpatrick's Dermatology, Ch. 17 (UV-induced DNA damage and NER, pp. 307-310)
This is a shared conversation. Sign in to Orris to start your own chat.