pathogenesis of membranous nephropathy
Reading File
Finding Sources
Searching PubMed
"membranous nephropathy"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Loading Image
Loading Image
I now have comprehensive information from multiple authoritative sources. Let me compile a thorough answer.
Pathogenesis of Membranous Nephropathy
Membranous nephropathy (MN) is characterized by the accumulation of immune complexes in the subepithelial space of the glomerular basement membrane (GBM), between podocyte foot processes and the GBM itself. Unlike many other glomerulonephritides, these deposits form primarily in situ rather than being pre-formed circulating complexes that passively deposit.
1. Experimental Foundation: Heymann Nephritis Model
Much of what is understood about MN pathogenesis came from the passive Heymann nephritis rat model, in which antibodies against megalin (a multiligand endocytic receptor on the basolateral surface of podocytes) create an identical histological picture. In this model, circulating antibodies cross the glomerular capillary wall, bind megalin in clathrin-coated pits on podocyte foot processes, and the resulting immune complexes are shed into the subepithelial space to form granular deposits. Megalin is not expressed in humans, but the model established the principle of in situ immune complex formation at the podocyte surface.
2. Primary (Autoimmune) MN: Podocyte Autoantigens
PLA2R - Phospholipase A2 Receptor (70-80% of primary MN)
The key discovery was that M-type phospholipase A2 receptor (PLA2R), a 180 kDa transmembrane glycoprotein expressed on the basolateral surface of podocytes, is the primary autoantigen in human primary MN.
- Circulating IgG4 anti-PLA2R autoantibodies are found in 70-80% of patients with primary MN
- PLA2R has a large extracellular domain with an N-terminal cysteine-rich domain (CysR), a fibronectin type II domain (FnII), and eight C-type lectin domains (CTLD1-8)
- Three reactive epitopes are targeted: CysR, CTLD1, and CTLD7
- High anti-PLA2R antibody activity is associated with intramolecular epitope spreading from CysR to CTLD1 and CTLD7 - and this spreading predicts decreased prospects of clinical remission
- A genome-wide association study (GWAS) found significant linkage to PLA2R1 on chromosome 2q24 and HLA-DQA1 on chromosome 6p21 - individuals homozygous for both risk variants have an odds ratio of nearly 80 for developing MN
THSD7A - Thrombospondin Type-1 Domain-Containing 7A (~5% of primary MN)
- Accounts for ~5% of primary MN in Western countries, higher prevalence in Japanese patients
- In contrast to PLA2R-positive MN, THSD7A-associated MN has a female predominance and is associated with malignancy
- Located on the basal surface of podocytes; like PLA2R, redistributes to form subepithelial immune deposits
Other Autoantigens (rare)
- NELL1 (neural EGF-like 1): IgG1-predominant autoantibodies; up to one-third of cases associated with malignancy
- Semaphorin 3B: found in some early childhood MN
- NEP (neutral endopeptidase): causes rare antenatal MN via alloimmunization - a genetically NEP-deficient mother transfers anti-NEP antibodies transplacentally
- Intracellular antigens including aldose reductase, superoxide dismutase 2, and alpha-enolase may contribute to podocyte injury
3. Mechanism of In Situ Immune Complex Formation
Two main mechanisms operate:
A. Intrinsic podocyte antigens (primary MN):
Circulating antibodies cross the glomerular capillary wall (which is permeable to IgG), bind to antigens (PLA2R, THSD7A) in clathrin-coated pits on the foot process basal surface, and the antigen-antibody complexes are shed into the subepithelial space.
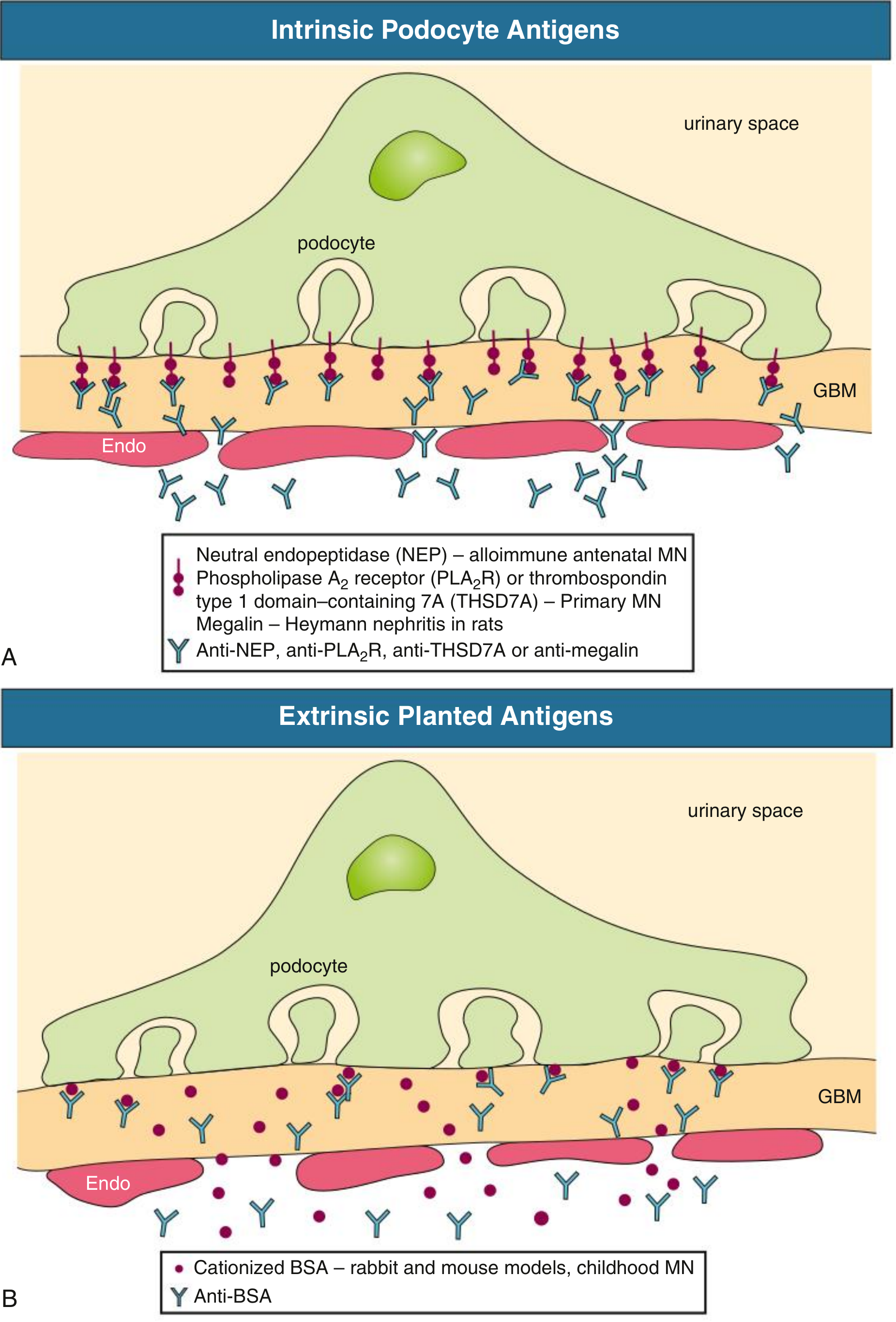
B. Extrinsic planted antigens (secondary MN):
- Cationized bovine serum albumin (cBSA) - documented in childhood MN from bottle milk exposure - plants in the GBM due to its positive charge, then binds circulating anti-BSA antibodies
- Tumor-shed antigens, viral antigens (HBV surface antigen), and drug haptens can similarly plant and react with circulating antibodies
- Immune complexes may also initially deposit subendothelially, then dissociate and reform in the subepithelial location
4. Downstream Effector Mechanisms: Complement and Podocyte Injury
Once subepithelial deposits form, the key effector pathway is complement activation, specifically generation of the membrane attack complex (C5b-9):
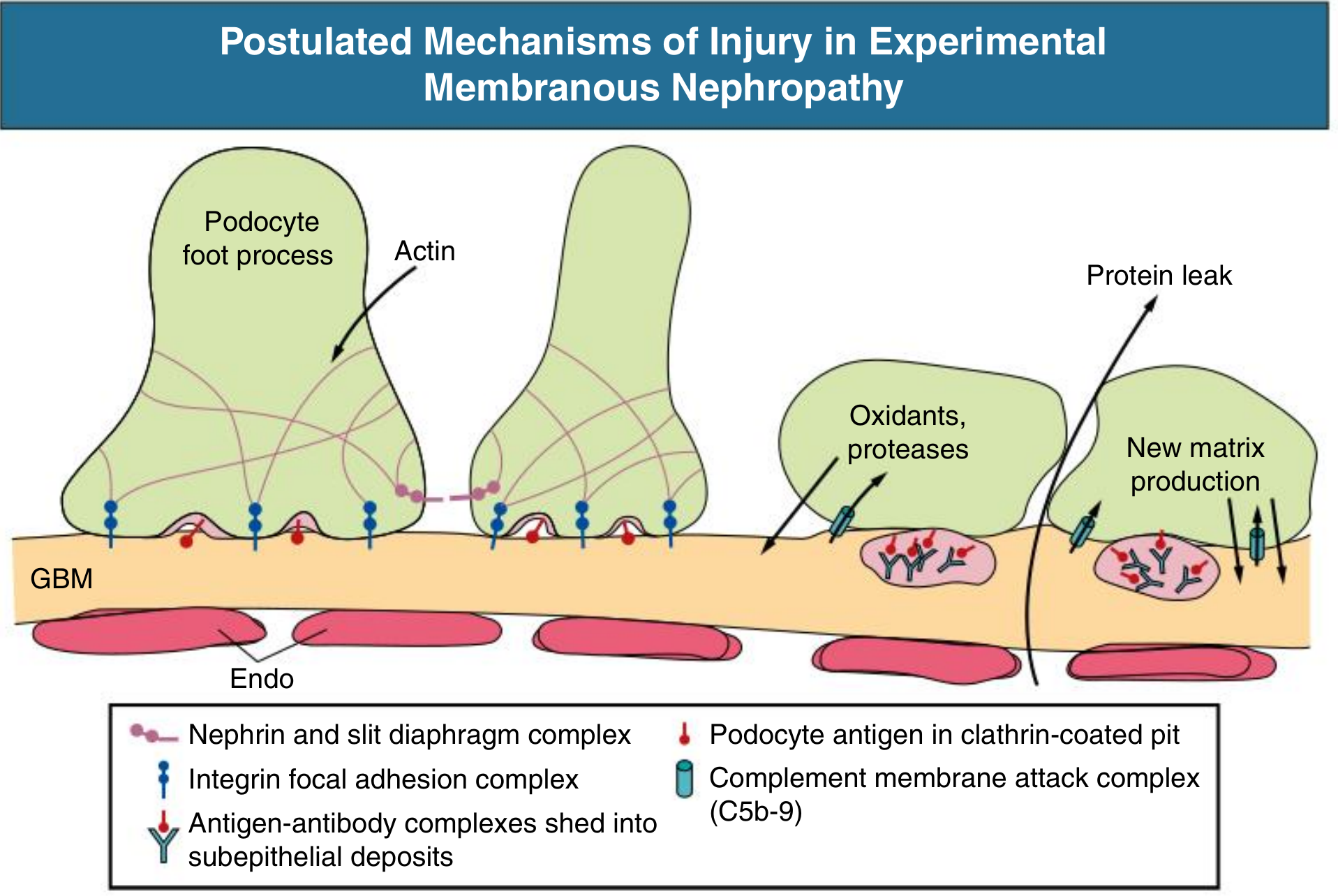
Key steps:
- Complement activation - The predominant antibody isotype in primary MN is IgG4, which is a poor activator of the classical complement pathway. The exact pathway of complement activation remains incompletely understood; the lectin pathway may be involved
- Sublytic C5b-9 insertion - The deposited C5b-9 does not cause cell lysis (the podocyte is too large) but instead acts as a sublytic stimulus, triggering the podocyte to release:
- Reactive oxygen species (oxidants)
- Proteases
- Prostaglandins and other lipid mediators
- Actin cytoskeleton disruption - These mediators disrupt actin polymerization in the foot processes, altering cell-matrix adhesion and displacing slit diaphragm proteins (nephrin, podocin)
- Foot process effacement - The result is loss of the charge-selective and size-selective filtration barrier, causing heavy proteinuria
- GBM remodeling - Damaged podocytes secrete new matrix material that expands the GBM around and between deposits. Over time this produces the characteristic spike-and-dome pattern on silver stain (the "spikes" are GBM matrix projections growing between deposits)
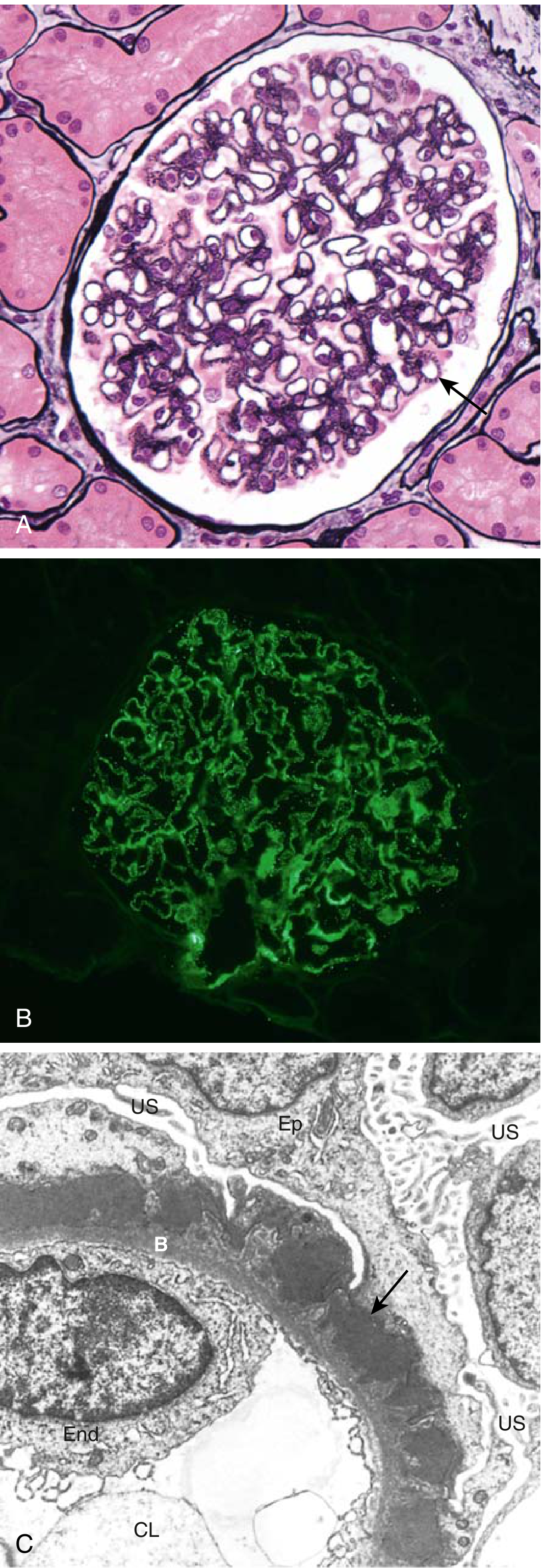
5. Morphological Staging (Ehrenreich-Churg)
The progressive GBM remodeling is captured in four EM stages:
| Stage | EM Findings |
|---|---|
| I | Small subepithelial deposits; no GBM spikes |
| II | Subepithelial deposits with adjacent GBM spikes |
| III | GBM matrix surrounds deposits (intramembranous) |
| IV | Distorted, thickened GBM; electron-lucent areas from deposit resorption |
On light microscopy: normocellular glomeruli with diffuse capillary wall thickening (no inflammation, no proliferation). On immunofluorescence: granular "lumpy-bumpy" IgG and C3 along the GBM. By immunofluorescence IgG subclass staining:
- Primary MN (PLA2R-associated): IgG4 dominant (IgG1 may dominate in early stages)
- Lupus membranous (class V LN): IgG1, IgG2, IgG3 predominate
- Malignancy-associated MN: IgG1, IgG2 predominate
6. Secondary MN Mechanisms
When MN is secondary to another condition, several mechanisms can generate subepithelial deposits:
| Cause | Mechanism |
|---|---|
| Hepatitis B | HBsAg or HBeAg as planted antigen; immune complex trapping |
| SLE (class V LN) | Autoantibodies including anti-dsDNA targeting subendothelial then subepithelial deposits |
| Malignancy | Tumor-shed antigens plant in GBM or form circulating complexes; altered immune surveillance |
| Drugs (penicillamine, gold) | Hapten-carrier mechanisms; altered self-antigen exposure |
| Syphilis/malaria | Microbial antigen-antibody complexes |
7. Role of Autoimmunity and Genetics
- Strong genetic association with HLA-DQA1 (class II HLA) - implicates aberrant CD4+ T cell help in tolerating anti-podocyte autoantibodies
- Risk allele in PLA2R1 likely lies in a regulatory region that may upregulate renal PLA2R expression, increasing antigen load
- B cell dysregulation is central - rituximab (anti-CD20) induces remission by depleting B cells and reducing anti-PLA2R titers
- Patients with PLA2R epitope spreading to CTLD1 and CTLD7 have high circulating titers and are less likely to respond to rituximab alone, pointing to deep B cell memory involvement
8. Disease Activity and Remission
Disease remains active as long as circulating autoantibodies persist. Immunologic remission (antibody disappearance) precedes clinical remission (proteinuria reduction) by weeks to months, because GBM remodeling lags behind antibody clearance. Once extensive podocyte loss occurs, proteinuria may persist even after immunologic remission, and progressive glomerulosclerosis follows.
Sources:
- Robbins & Kumar Basic Pathology - Chapter 12
- Comprehensive Clinical Nephrology, 7th Ed. - Chapter 21
- Brenner and Rector's The Kidney, 2-Volume Set - Chapter 17
- Henry's Clinical Diagnosis and Management by Laboratory Methods - Chapter 55
This is a shared conversation. Sign in to Orris to start your own chat.