Gout for mbbs 1st yr mbbs
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Loading Image
I now have comprehensive, well-sourced content from Robbins Basic Pathology, Lippincott Biochemistry, and Lippincott Pharmacology. I have all the information needed for a thorough 1st year MBBS gout note. Let me compile the complete answer.
GOUT - Complete Notes for 1st Year MBBS
Sources: Robbins & Kumar Basic Pathology; Lippincott Illustrated Reviews: Biochemistry 8e; Lippincott Illustrated Reviews: Pharmacology
1. Definition
Gout is a metabolic disorder characterized by:
- Hyperuricemia (elevated uric acid in blood, >6.8 mg/dL)
- Deposition of monosodium urate (MSU) crystals in joints and soft tissues
- Recurrent attacks of acute arthritis, and in chronic cases, formation of tophi
Key rule: Hyperuricemia is necessary but not sufficient for gout. Gout is always preceded by hyperuricemia, but only ~10% of hyperuricemic individuals develop gout.
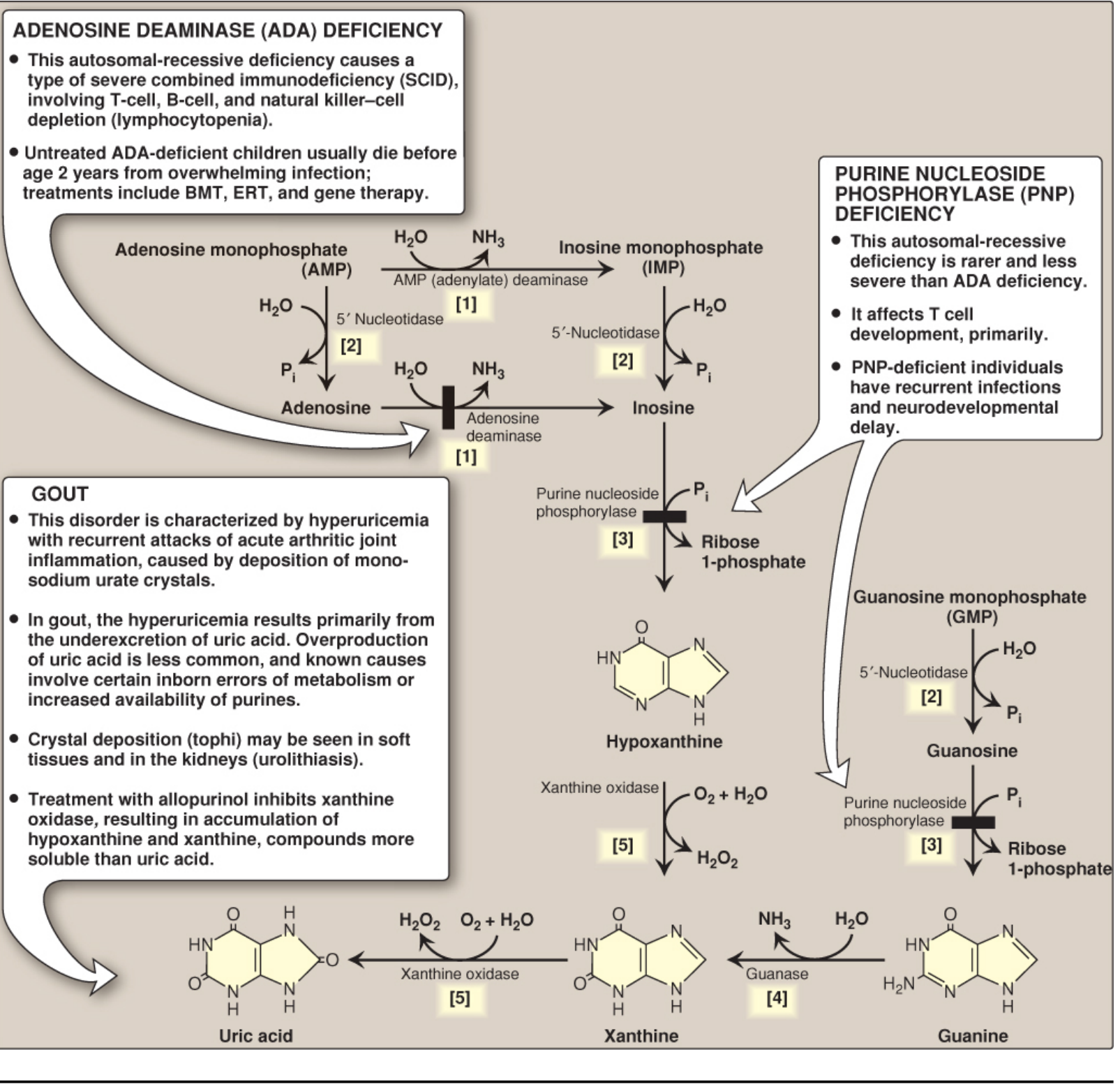
2. Biochemical Basis - Purine Degradation Pathway
Uric acid is the end product of purine catabolism in humans. The pathway:
AMP / GMP → Nucleosides → Hypoxanthine / Guanine → Xanthine → Uric Acid
Key enzyme: Xanthine oxidase (XO) - a molybdenum-containing enzyme that oxidizes:
- Hypoxanthine → Xanthine
- Xanthine → Uric acid
Uric acid is excreted primarily in the urine.
3. Causes of Hyperuricemia
A. Underexcretion (>90% of cases)
- Primary: Idiopathic defect in renal urate excretion (most common)
- Secondary:
- Drugs: thiazide diuretics, low-dose aspirin, cyclosporine
- Lead poisoning (saturnine gout)
- Lactic acidosis (lactate competes with urate for excretion)
- Chronic kidney disease
B. Overproduction (<10% of cases)
- Primary (idiopathic) - most common in this category
- Enzyme defects:
- PRPP synthetase overactivity - increased purine synthesis
- HGPRT (hypoxanthine-guanine phosphoribosyltransferase) deficiency - interrupts the purine salvage pathway; partial deficiency → gout; complete absence → Lesch-Nyhan syndrome (hyperuricemia + self-mutilation + neurologic features)
- Secondary overproduction:
- Myeloproliferative disorders (high cell turnover)
- Chemotherapy / tumor lysis syndrome
- Von Gierke disease (glycogen storage disorder)
Risk factors for developing gout (from hyperuricemia):
- Age and duration of hyperuricemia (typically 20-30 years before gout appears)
- Male sex
- Diet rich in meat, shellfish, alcohol (especially beer)
- Diet poor in low-fat dairy (dairy products are protective)
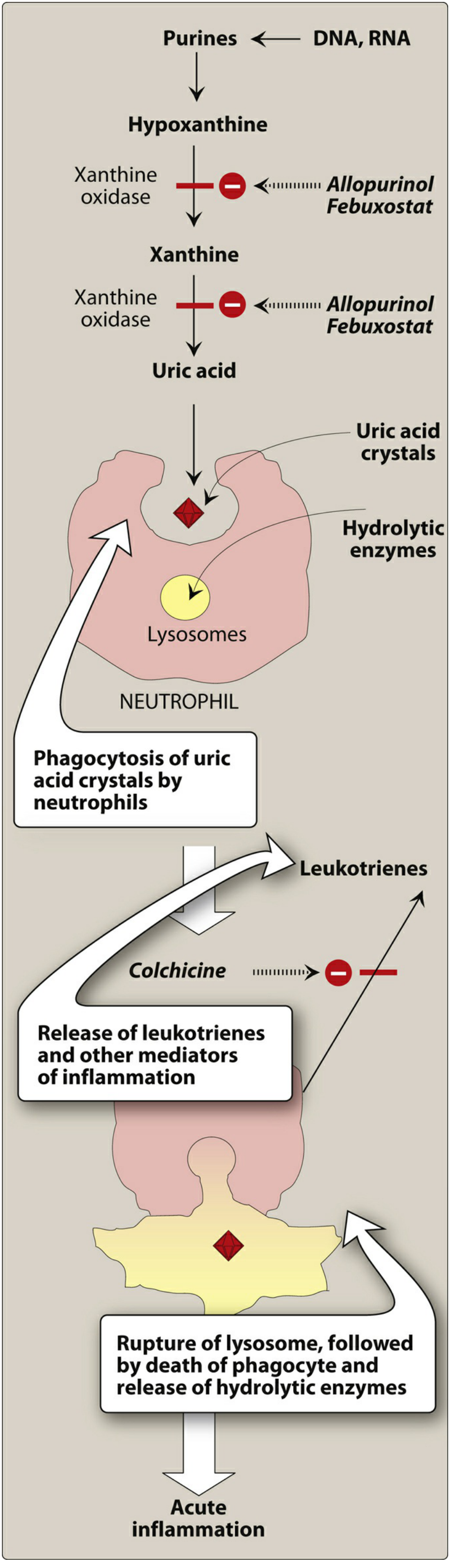
4. Pathogenesis of Joint Inflammation
The mechanism of acute gouty arthritis:
- MSU crystals precipitate in the joint
- Resident synovial macrophages phagocytose crystals → activates the NLRP3 inflammasome
- Inflammasome activates caspase-1 → produces active IL-1β
- IL-1β recruits neutrophils into the joint
- Neutrophils also phagocytose crystals → lysosomal membranes rupture → hydrolytic enzymes released
- Release of leukotrienes, free radicals, and proteases → acute arthritis
- Attack typically remits spontaneously in days to weeks
With repeated attacks:
- Formation of tophi (aggregates of MSU crystals + inflammatory tissue) in synovium and periarticular tissue
- Cartilage damage, joint destruction
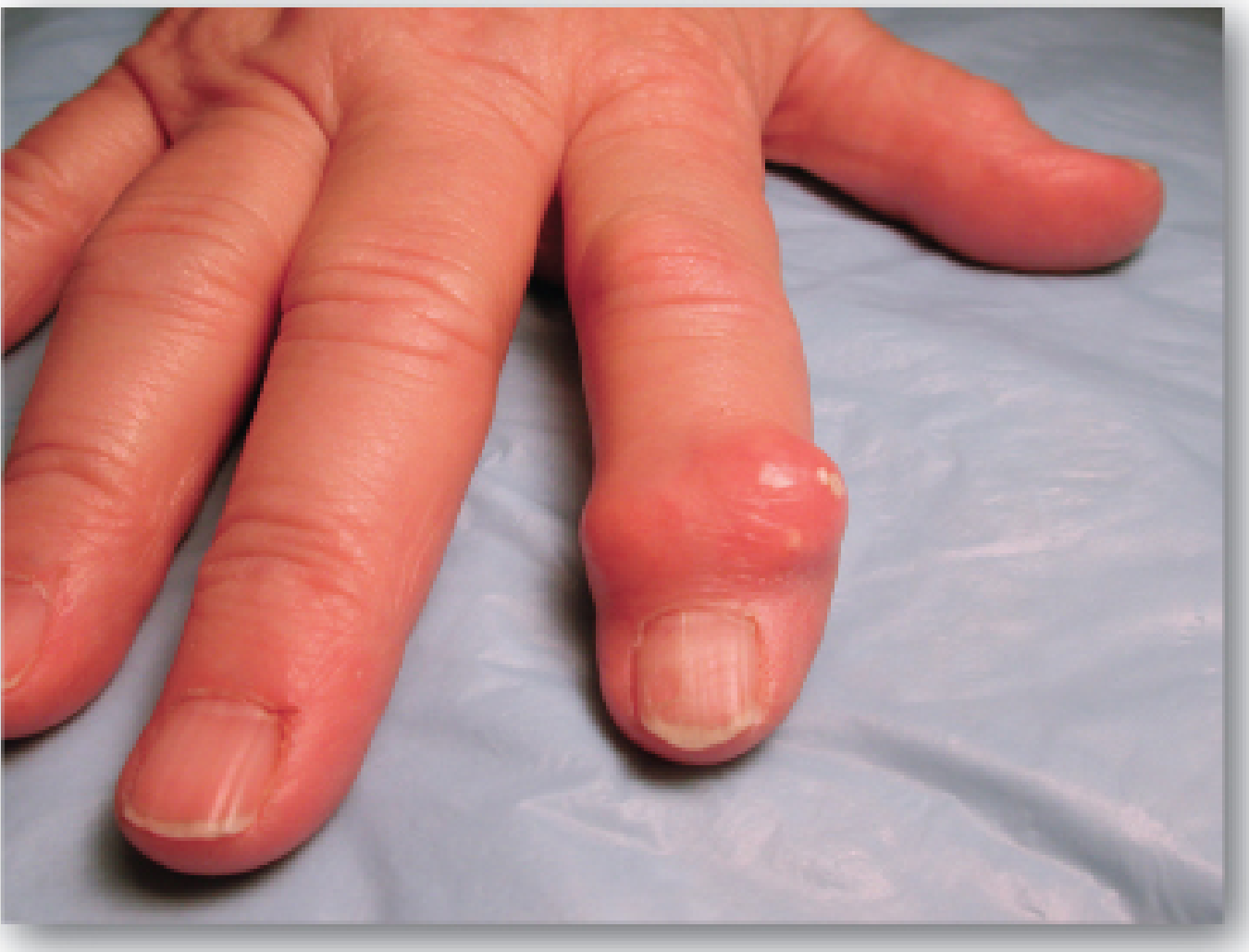
5. Clinical Features (Four Stages)
| Stage | Description |
|---|---|
| 1. Asymptomatic hyperuricemia | Elevated uric acid (>6.8 mg/dL), no symptoms. May last 20-30 years |
| 2. Acute gouty arthritis | Sudden, severe pain, swelling, redness and warmth in a joint. Classic site: 1st MTP joint (big toe) = podagra. Also affects ankles, knees, wrists, elbows |
| 3. Intercritical gout | Symptom-free intervals between attacks |
| 4. Chronic tophaceous gout | Persistent joint disease with tophi deposits in soft tissues, cartilage, tendons, kidney |
6. Complications
- Urolithiasis - uric acid kidney stones
- Gouty nephropathy - urate crystal deposition in renal tubules/interstitium
- Joint destruction in chronic disease
7. Diagnosis
Definitive diagnosis: Aspiration of synovial fluid from affected joint → polarized light microscopy
- Needle-shaped MSU crystals with negative birefringence (yellow when parallel to the polarizer axis)
Serum uric acid: >6.8 mg/dL (may be normal during acute attack)
8. Treatment
A. Acute Attack
| Drug | Mechanism | Notes |
|---|---|---|
| NSAIDs (Indomethacin) | Inhibit COX → reduce prostaglandin synthesis | Drug of choice for acute attack |
| Colchicine | Binds tubulin → depolymerizes microtubules → blocks neutrophil migration into joint | Must give within 36 hours of onset; relieves pain in 12 hours |
| Corticosteroids | Anti-inflammatory | Used when NSAIDs/colchicine contraindicated; intra-articular or systemic |
B. Chronic Gout / Urate-Lowering Therapy (ULT)
Indications for ULT: >2 attacks/year, chronic kidney disease, kidney stones, or tophi
Goal: Reduce serum urate below 6 mg/dL (saturation point)
Xanthine Oxidase Inhibitors (First-line ULT)
| Drug | Details |
|---|---|
| Allopurinol | Purine analog; competitively inhibits xanthine oxidase (last 2 steps of uric acid synthesis); first-line preferred over febuxostat; adverse effect: hypersensitivity/skin rash (more common in renal impairment) |
| Febuxostat | Non-purine XO inhibitor; less renal elimination than allopurinol; reserved for patients intolerant to allopurinol; caution in heart disease/stroke history |
Uricosuric Agents (for underexcretors)
| Drug | Mechanism | Notes |
|---|---|---|
| Probenecid | Inhibits urate-anion exchanger in proximal tubule → blocks urate reabsorption → increases uric acid excretion | Avoid if creatinine clearance <50 mL/min |
Recombinant Uricase
| Drug | Mechanism | Notes |
|---|---|---|
| Pegloticase | Recombinant urate oxidase; converts uric acid to allantoin (water-soluble) | IV infusion every 2 weeks; for refractory gout; risk of anaphylaxis |
Important Note: Starting ULT can precipitate an acute attack (due to rapid shifts in serum urate). Co-prescribe low-dose colchicine or NSAIDs for at least 6 months when initiating ULT.
9. Colchicine - Detailed Pharmacology (High-Yield)
- Source: Plant alkaloid
- MOA: Binds tubulin → depolymerizes microtubules → blocks neutrophil migration + inhibits cell division (blocks mitotic spindle)
- NOT uricosuric, NOT analgesic
- Pharmacokinetics: Oral; absorbed from GI tract; metabolized by hepatic CYP3A4; enterohepatic recirculation
- Adverse effects: Nausea, vomiting, diarrhea, abdominal pain (most common); chronic use → myopathy, neutropenia, aplastic anemia, alopecia
- Interactions: CYP3A4 inhibitors (clarithromycin, itraconazole) and P-gp inhibitors (amiodarone, verapamil) increase toxicity
- Contraindicated in pregnancy
10. High-Yield Summary Table
| Feature | Key Fact |
|---|---|
| Crystal type | Monosodium urate (MSU) |
| Crystal shape | Needle-shaped, negative birefringence |
| Diagnosis | Polarized light microscopy of synovial fluid |
| Most common cause | Underexcretion (>90%) |
| Enzyme in crystal inflammation | Xanthine oxidase, NLRP3 inflammasome, caspase-1, IL-1β |
| Classic site | 1st MTP joint (podagra) |
| Acute treatment | NSAIDs (indomethacin) > Colchicine > Steroids |
| Chronic treatment | Allopurinol (1st line), Febuxostat, Probenecid |
| Lesch-Nyhan | Complete HGPRT deficiency → gout + neurologic features |
| Saturnine gout | Lead poisoning |
Sources:
- Robbins & Kumar Basic Pathology (Robbins Pathology), Chapter 19 - Bones, Joints and Soft Tissues
- Lippincott Illustrated Reviews: Biochemistry 8th ed, Chapter 22 - Purine Degradation
- Lippincott Illustrated Reviews: Pharmacology, Chapter 40 - Drugs for Gout
Glycogen storage disease chart
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
I now have all the content needed - comprehensive data from Harper's Biochemistry (full table with Types 0-VIII), Robbins Cotran (detailed clinical table), and two excellent diagrams. Let me compile the complete GSD chart.
Glycogen Storage Diseases (Glycogenoses) - Complete Chart
Sources: Robbins & Cotran Pathologic Basis of Disease; Harper's Illustrated Biochemistry 32nd Ed; Lippincott Biochemistry 8th Ed
Glycogen Pathway - Sites of Enzyme Defects
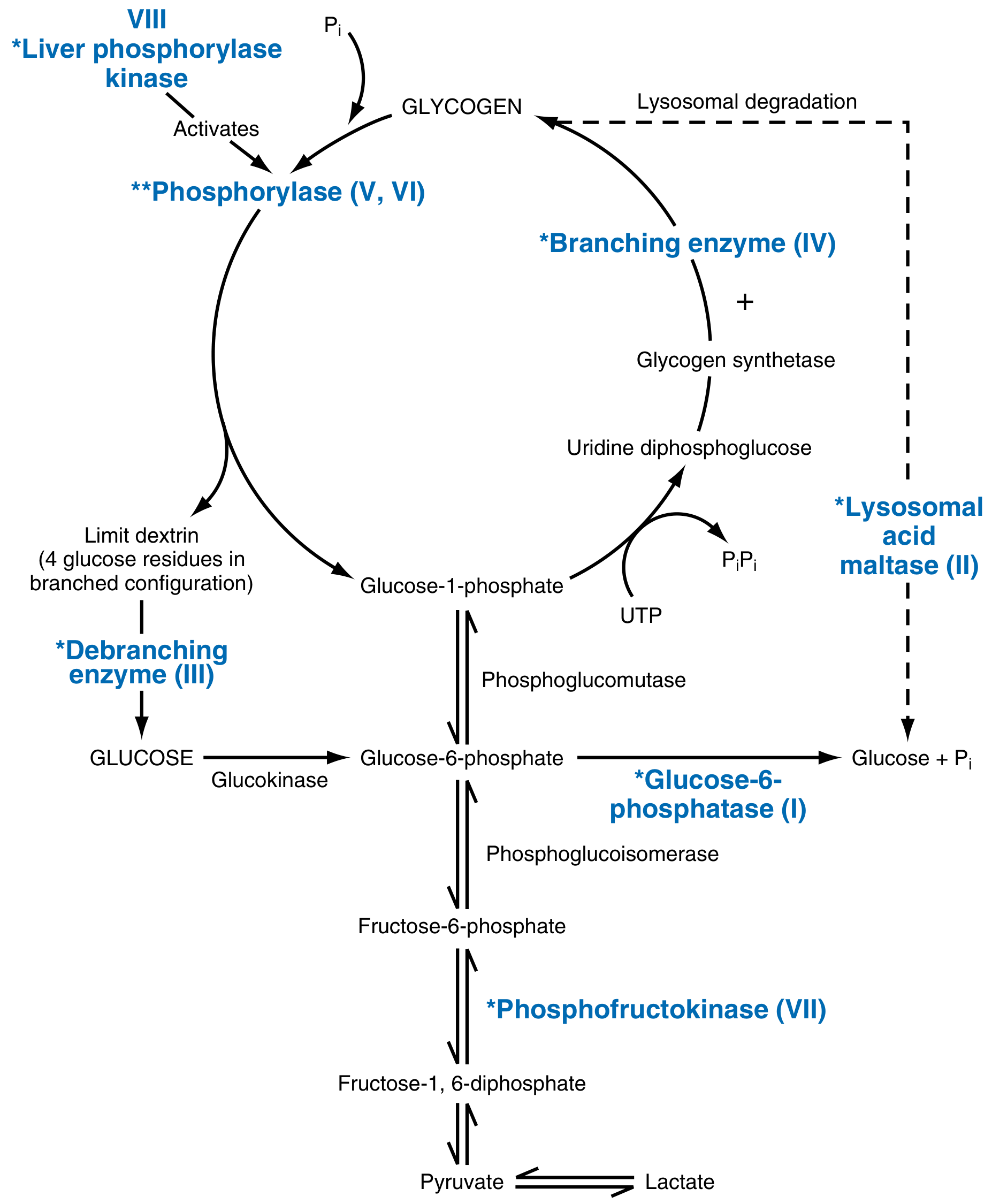
Hepatic vs Myopathic GSD - Visual Summary
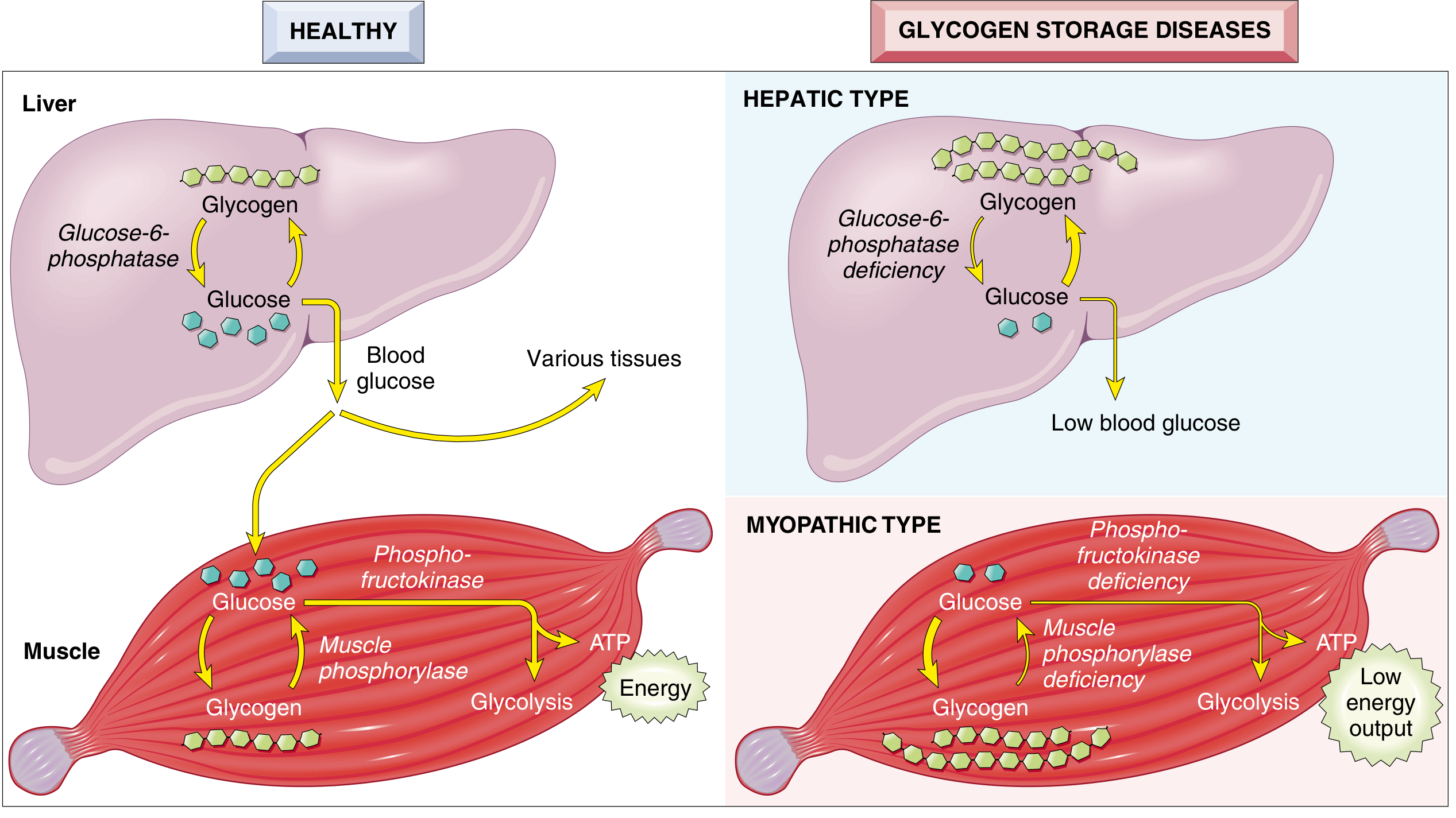
Master GSD Chart
| Type | Eponym | Enzyme Deficiency | Gene | Organs Affected | Key Clinical Features |
|---|---|---|---|---|---|
| 0 | - | Glycogen synthase (GYS2) | GYS2/GYS1 | Liver | Hypoglycemia, hyperketonemia, early death; cannot store glycogen |
| I (Ia) | Von Gierke disease | Glucose-6-phosphatase | G6PC | Liver, kidney | Hypoglycemia (severe, fasting), hepatomegaly, renomegaly, lactic acidemia, hyperlipidemia, hyperuricemia → gout, skin xanthomas, bleeding tendency, stunted growth |
| I (Ib) | Von Gierke (variant) | G6P transporter (ER membrane) | SLC37A4 | Liver, kidney | Same as Ia + neutropenia, recurrent infections |
| II | Pompe disease | Lysosomal acid α-glucosidase (acid maltase) | GAA | All organs (especially heart + muscle) | Massive cardiomegaly, hypotonia, cardiorespiratory failure by age 2 (infantile); Adult form = chronic myopathy only. Only lysosomal GSD. Enzyme replacement therapy available |
| III | Cori disease / Forbes disease | Debranching enzyme (amylo-1,6-glucosidase) | AGL | Liver + muscle | Fasting hypoglycemia, hepatomegaly, accumulation of limit dextrin (abnormal branched glycogen), variable muscle weakness |
| IV | Andersen disease (Amylopectinosis) | Branching enzyme | GBE1 | Liver, heart, all tissues | Accumulation of amylopectin-like polysaccharide (few branch points), progressive cirrhosis, heart or liver failure; often fatal in childhood |
| V | McArdle disease | Muscle phosphorylase (myophosphorylase) | PYGM | Skeletal muscle only | Painful cramps on exercise, myoglobinuria (50%), no rise in blood lactate after exercise (block in glycolysis), onset >20 years, elevated serum CK, normal longevity |
| VI | Hers disease | Liver phosphorylase | PYGL | Liver | Hepatomegaly, mild hypoglycemia, generally benign course |
| VII | Tarui disease | Muscle phosphofructokinase (PFK) | PFKM | Skeletal muscle, RBCs | Similar to McArdle (exercise intolerance, cramps, myoglobinuria); also hemolytic anemia (RBC PFK also deficient) |
| VIII / IX | - | Phosphorylase kinase | PHKA2 | Liver | Hepatomegaly, mild hypoglycemia; usually benign, X-linked form common |
Three Pathophysiological Groups
| Group | Mechanism | Key Diseases | Main Problem |
|---|---|---|---|
| Hepatic type | Defective glycogen breakdown in liver → cannot release glucose to blood | Type I (Von Gierke), Type III (Cori), Type VI (Hers) | Hypoglycemia + hepatomegaly |
| Myopathic type | Defective glycolysis in muscle → cannot generate ATP for exercise | Type V (McArdle), Type VII (Tarui) | Exercise intolerance, cramps, no lactate rise |
| Generalized / Miscellaneous | Affects all organs or doesn't fit above categories | Type II (Pompe), Type IV (Andersen) | Multi-organ failure (heart dominant in Pompe) |
High-Yield Memory Tricks
| Mnemonic | Meaning |
|---|---|
| "Very Poor Carbohydrate Metabolism Always Has Terrible Results" | Von Gierke, Pompe, Cori, McArdle, Andersen, Hers, Tarui |
| Types I, III, VI, IX = Hepatic (hypoglycemia) | All affect liver phosphoglucose release |
| Types V, VII = Myopathic (exercise-related) | Muscle enzymes, no lactate rise on exercise |
| Type II = "2 problems" | Lysosomal + cytoplasmic glycogen accumulation; the only GSD that is also a lysosomal storage disease |
| Von Gierke = "can't go" | Cannot release glucose → severe fasting hypoglycemia |
| McArdle = "muscle ache" | Muscle cramps + myoglobinuria on exertion |
| Pompe = "pump failure" | Cardiac pump fails → cardiomegaly + death by age 2 |
| Andersen = "abnormal branching" | Defective branching enzyme → amylopectin-like glycogen |
Key High-Yield Facts for Exams
- Most common GSD: Type I (Von Gierke)
- Only lysosomal GSD: Type II (Pompe) - enzyme is in lysosomes
- Only GSD with cardiomegaly as dominant feature: Pompe
- No lactate rise on exercise: Myopathic types (McArdle, Tarui) - block in glycolysis
- Limit dextrin accumulation: Type III (Cori) - debranching enzyme defect
- Inheritance: All types are autosomal recessive, except Type IX (X-linked phosphorylase kinase deficiency)
- Treatment available: Pompe disease - enzyme replacement therapy (alglucosidase alfa)
- GSD + gout + xanthomas: Von Gierke (Type I) - secondary hyperuricemia and hyperlipidemia
- GSD + hemolytic anemia: Type VII (Tarui) - PFK deficiency in RBCs too
Sources: Robbins & Cotran Pathologic Basis of Disease (Table 5.7); Harper's Illustrated Biochemistry 32nd Ed (Table 18-2); Lippincott Biochemistry 8th Ed (Chapter 11)
Pem for biochemistry 1st yr mbbs student
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
Finding Sources
Reading File
I now have comprehensive content from all the key textbooks. Let me compile the full PEM notes for 1st year MBBS biochemistry.
Protein-Energy Malnutrition (PEM) - Biochemistry Notes for 1st Year MBBS
Sources: Lippincott Illustrated Reviews: Biochemistry 8e; Park's Textbook of Preventive & Social Medicine; Sleisenger & Fordtran's GI & Liver Disease
1. Definition
Protein-Energy Malnutrition (PEM) is a spectrum of nutritional disorders resulting from inadequate intake of protein and/or calories, leading to a range of clinical syndromes - from mild growth retardation to severe life-threatening states.
- Also called Protein-Energy Undernutrition (PEU)
- Most prevalent nutritional deficiency worldwide
- Predominantly affects children under 5 years in developing countries
2. Classification of PEM
A. By Clinical Form
| Feature | Kwashiorkor | Marasmus | Marasmic-Kwashiorkor |
|---|---|---|---|
| Primary deficit | Protein >> Calories | Calories + Protein both | Both forms combined |
| Weight for age | 60-80% of expected | <60% of expected | <60% + edema |
| Weight for height | Normal or ↓ | Markedly ↓ | Markedly ↓ |
| Edema | Present (pitting) | Absent | Present |
| Muscle wasting | Present (masked by edema) | Severe | Severe |
| Subcutaneous fat | Preserved | Markedly depleted | Depleted |
| Serum albumin | Markedly ↓ | Low/normal | Low |
| Fatty liver | Present | Absent | Variable |
| Hair changes | Depigmented, sparse, flag sign | Sparse | Variable |
| Skin changes | Flaky paint, dermatitis | Loose, wrinkled | Variable |
| Appetite | Poor (anorexia) | Often preserved | Poor |
| Mood | Miserable, irritable, apathetic | Alert (anxious look) | Mixed |
| Age | Post-weaning (~1-3 yrs) | Infants <1 yr | Any |
| Insulin levels | Relatively maintained (CHO diet) | Very low | Low |
| Adaptation type | Nonadapted malnutrition | Adapted malnutrition | Mixed |
B. Waterlow Classification (by Wasting and Stunting)
| Category | Wasting (Wt/Ht) | Stunting (Ht/Age) | Interpretation |
|---|---|---|---|
| Normal | >90% | >95% | Normal |
| Stunting only | >90% | <95% | Chronic, past malnutrition |
| Wasting only | <90% | >95% | Acute, current malnutrition |
| Wasting + Stunting | <90% | <95% | Acute-on-chronic malnutrition |
C. Gomez Classification (by Weight for Age)
| Grade | % Expected Weight | Severity |
|---|---|---|
| Normal | >90% | Normal |
| Grade I | 75-90% | Mild |
| Grade II | 60-75% | Moderate |
| Grade III | <60% | Severe |
3. Kwashiorkor - Detailed
Meaning
The word "kwashiorkor" comes from the Ga language of West Africa - means "disease of the displaced child" (child displaced from the breast when the next sibling is born).
Cause
- Protein deprivation relatively greater than calorie deprivation
- Diet rich in carbohydrates but poor in protein (e.g., maize-based diet after weaning)
- Often precipitated by an acute infection or physiological stress on a chronically malnourished child
Biochemical Basis
- Low protein intake → severely decreased synthesis of visceral proteins (albumin, transferrin, prealbumin)
- ↓ Serum albumin → reduced plasma oncotic pressure → edema (fluid shifts from intravascular to interstitial compartment)
- Adequate carbohydrate intake → insulin levels maintained → lipolysis and proteolysis suppressed
- Fat cannot be mobilized from liver → fatty liver (hepatic steatosis)
- Called "nonadapted malnutrition" because the body cannot properly adapt to protein lack
Clinical Features
- Bilateral pitting edema (legs, feet, face - "moon face")
- Protuberant abdomen (edema + hepatomegaly + weak abdominal muscles)
- Fatty liver / hepatomegaly
- Skin changes: "flaky paint" dermatosis, hyperpigmented patches, areas of peeling
- Hair changes: sparse, depigmented, easily pluckable, "flag sign" (bands of light/dark hair)
- Growth retardation (stunted)
- Apathy, lethargy; irritable when handled
- Depressed immunity → infections common
4. Marasmus - Detailed
Cause
- Calorie deprivation relatively greater than protein deprivation
- Both calories AND protein are deficient
- Usually in infants <1 year when breast milk is replaced with watery gruels
Biochemical Basis
- ↓↓ Caloric intake → insulin levels fall markedly
- ↓ Insulin → lipolysis activated → fat stores mobilized
- Gluconeogenesis activated → muscle protein broken down for energy
- Body adapts by reducing metabolic rate, mobilizing all energy stores
- Called "adapted malnutrition" - the body has adapted to starvation
Clinical Features
- Severe wasting - "skin and bones," "old man face" (wrinkled face)
- No edema (distinguishes from kwashiorkor)
- Markedly depleted subcutaneous fat and muscle
- Weight <60% of expected
- Marked muscle wasting - "baggy pants" appearance (redundant skin over buttocks)
- Weakness, severe growth retardation
- Serum albumin relatively preserved (no edema)
- No fatty liver
- Alert, anxious expression (but weak)
- Thin, sparse hair (but not depigmented)
Clinical Images
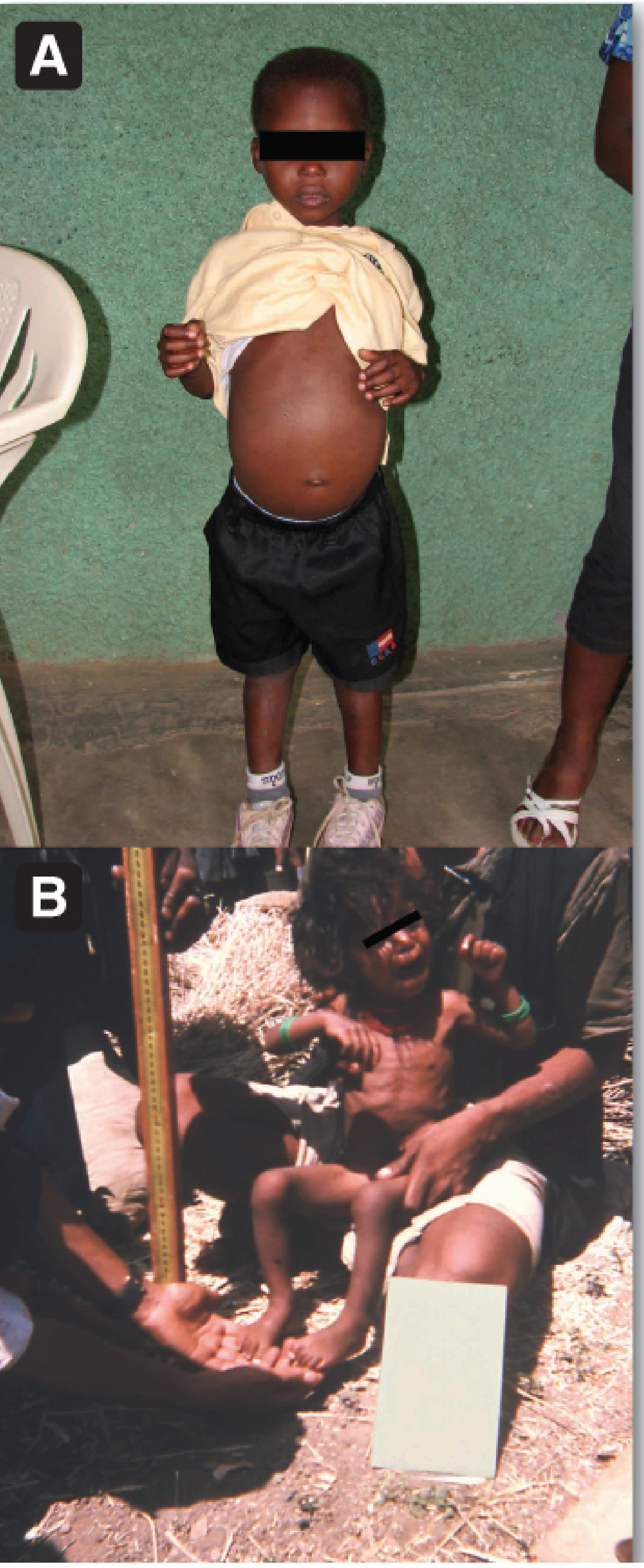
(Lippincott Biochemistry 8e - Fig 27.19: A = Kwashiorkor (swollen belly, edema); B = Marasmus (severe wasting/emaciation))
5. Biochemical / Metabolic Changes in PEM
| Parameter | Kwashiorkor | Marasmus |
|---|---|---|
| Serum albumin | Markedly ↓ (<2.8 g/dL) | Near normal or mildly ↓ |
| Serum transferrin | ↓ | Less affected |
| Serum prealbumin | ↓ | Less affected |
| Insulin | Relatively maintained | Markedly ↓ |
| Cortisol | ↑ (stress response) | ↑ |
| Glucagon | ↑ | ↑ |
| Blood glucose | May be normal | Low (hypoglycemia possible) |
| Plasma FFA | ↓ (lipolysis suppressed) | ↑ (lipolysis active) |
| Liver fat | ↑ (fatty liver) | Normal |
| Muscle mass | ↓ | ↓↓ (severe) |
| Immune function | ↓↓ | ↓ |
| GI function | Villous atrophy, ↓ enzymes | Villous atrophy |
6. Why Edema in Kwashiorkor but NOT in Marasmus?
This is a classic MBBS exam question:
- In kwashiorkor: ↓ protein synthesis → ↓ serum albumin → ↓ plasma oncotic pressure → fluid shifts to interstitial space → pitting edema
- In marasmus: Protein intake is also reduced but the body adapts; edema does NOT develop because the reduction is proportionate and albumin is relatively preserved
- Key rule: Edema in PEM = think Kwashiorkor
7. Comparison: Why Does Kwashiorkor Develop Fatty Liver?
| Step | Explanation |
|---|---|
| ↓ Protein intake | ↓ Synthesis of apolipoprotein B (apoB) - the protein component of VLDL |
| Cannot form VLDL | Fat cannot be exported from hepatocytes |
| Fat accumulates in liver | → Hepatic steatosis (fatty liver) |
| Adequate CHO intake | → Insulin maintained → de novo lipogenesis continues |
| Net result | Fat enters liver but cannot leave → fatty liver |
8. Assessment of PEM
| Tool | Parameter Measured | Significance |
|---|---|---|
| Weight for age | Underweight | Screening tool; affected by acute + chronic malnutrition |
| Height for age | Stunting | Reflects chronic malnutrition |
| Weight for height | Wasting | Reflects acute/current malnutrition |
| MUAC (Mid-upper arm circumference) | Muscle mass | >13.5 cm = normal; 12.5-13.5 = mild-moderate; <12.5 = severe |
| Serum albumin | Visceral protein stores | <3.5 g/dL = mild; <2.8 = severe |
| Serum prealbumin | Short-term protein status | Half-life ~2 days; sensitive early marker |
| Serum transferrin | Protein status | Half-life ~8 days |
| Skin fold thickness | Fat stores | Triceps skin fold |
9. Treatment Principles
Acute Phase (Stabilization)
- Treat hypoglycemia, hypothermia, dehydration
- Treat infections (broad-spectrum antibiotics)
- Correct electrolyte imbalances (K⁺, Mg²⁺, phosphate)
- Start feeding cautiously (avoid refeeding syndrome)
Refeeding syndrome warning: Rapid refeeding of carbohydrates → insulin surge → drives phosphate, K⁺, Mg²⁺ intracellularly → hypophosphatemia (most dangerous), hypokalemia, hypomagnesemia → cardiac arrhythmias, respiratory failure. Milk is given as it is rich in phosphate.
Rehabilitation Phase
- Gradually increase protein and calorie intake
- High-protein, high-calorie diet (milk, eggs, pulses)
- Treat micronutrient deficiencies (Vitamin A, zinc, iron)
- Treat underlying infections and parasites
Community Level Prevention (Park's)
- Promote breastfeeding
- Develop low-cost weaning foods
- Nutrition education
- Immunization (prevents infections that precipitate PEM)
- Family planning (birth spacing)
- Food fortification
10. High-Yield Summary Table
| Feature | Kwashiorkor | Marasmus |
|---|---|---|
| Meaning | "Displaced child" (Ga language) | Greek: "to waste away" |
| Primary deficiency | Protein >> calories | Calories + protein both |
| Age | Post-weaning (1-3 years) | Infancy (<1 year) |
| Edema | YES (hallmark) | No |
| Fatty liver | YES | No |
| Albumin | Markedly low | Near normal |
| Subcutaneous fat | Preserved | Absent |
| Skin/hair changes | Present (depigmentation) | Less prominent |
| Adaptation | Nonadapted | Adapted |
| Mood | Apathetic/miserable | Alert/anxious |
| Insulin levels | Relatively maintained | Low |
11. Mnemonics
| Mnemonic | Meaning |
|---|---|
| FRESH (Kwashiorkor) | Fatty liver, Retarded growth, Edema, Skin/hair changes, Hypoalbuminemia |
| SAME (Marasmus) | Skin and bones, Albumen near normal, Muscle wasting, Empty fat stores |
| Kwashiorkor = K for K | Kwashi + edema because low protein = low albumin = water can't stay in vessels |
| Marasmus = M for Meager | Everything is meager - calories AND protein, total starvation |
Sources:
- Lippincott Illustrated Reviews: Biochemistry 8th Ed, Chapter 27
- Park's Textbook of Preventive & Social Medicine
- Sleisenger & Fordtran's Gastrointestinal & Liver Disease
This is a shared conversation. Sign in to Orris to start your own chat.